🧬 Neurofibroma — Actualización 2026: espectro clínico-patológico, asociación con NF1 y manejo actualizado
El neurofibroma es un tumor benigno de la vaina nerviosa periférica, originado en una mezcla heterogénea de células de Schwann, fibroblastos, pericitos y células inflamatorias, a diferencia del schwannoma que es puramente de células de Schwann. Aunque puede presentarse como una lesión solitaria esporádica, su importancia clínica radica en su asociación con la neurofibromatosis tipo 1 (NF1) (enfermedad de von Recklinghausen), donde aparece en sus formas múltiple y plexiforme, esta última con riesgo de transformación maligna a sarcoma de la vaina nerviosa periférica (MPNST) del 8-12% a lo largo de la vida. La clasificación de la Organización Mundial de la Salud (OMS 2020) distingue tres variantes principales: neurofibroma localizado, neurofibroma difuso y neurofibroma plexiforme, este último patognomónico de NF1. Esta ficha, actualizada a 2026, desarrolla de manera extensa la epidemiología, clínica, radiología (con especial énfasis en el signo de la diana en RM), histopatología, inmunohistoquímica, genética molecular (pérdida de NF1, alteraciones en CDKN2A, TP53, SUZ12), diagnóstico diferencial, opciones terapéuticas (incluyendo los inhibidores de MEK como selumetinib en neurofibromas plexiformes) y el seguimiento oncológico prolongado.
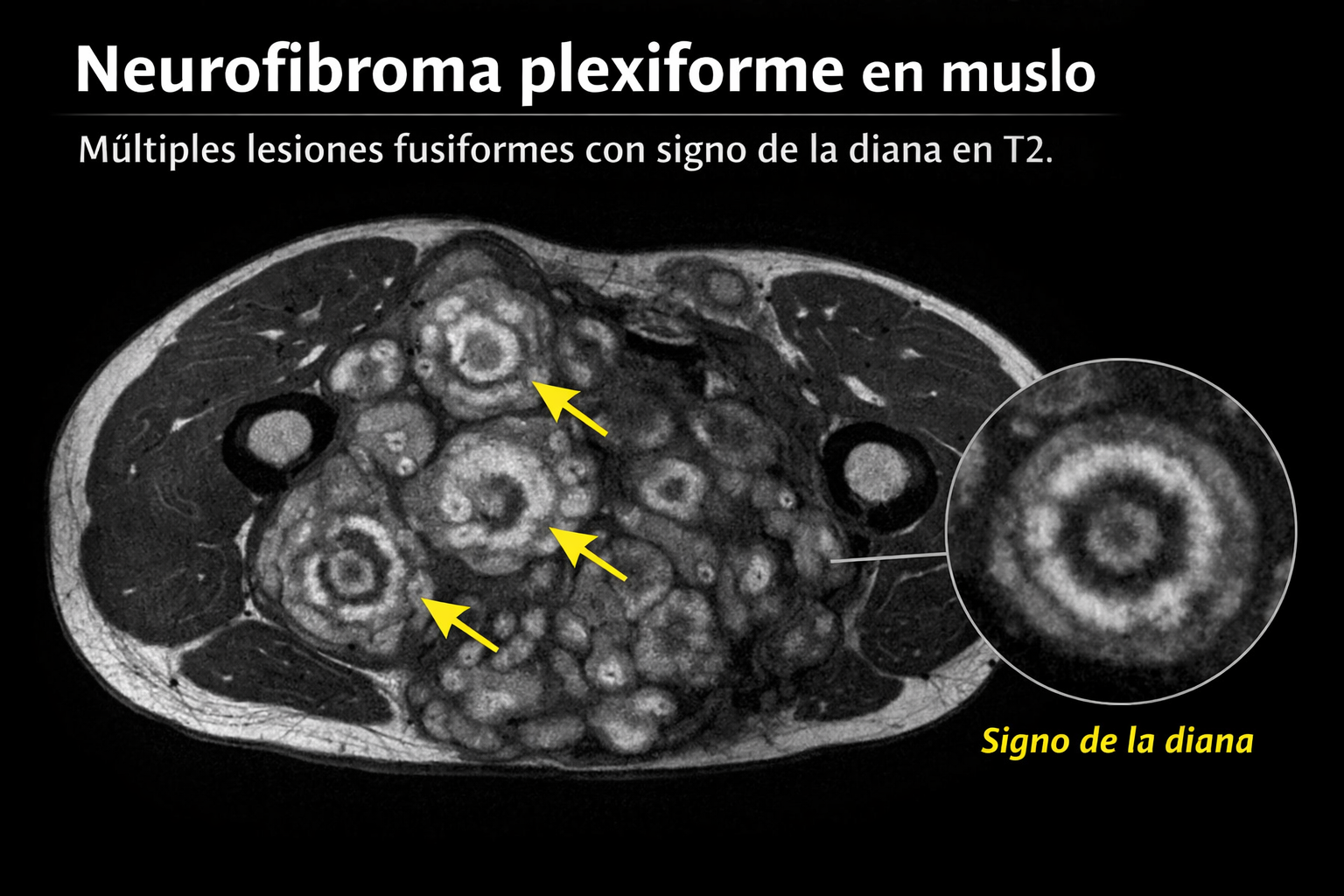
Hallazgo radiológico característico
0) En 1 minuto
- ✅ Definición: El neurofibroma es un tumor benigno de la vaina nerviosa periférica, no encapsulado, compuesto por una mezcla de células de Schwann, fibroblastos, pericitos, mastocitos y células inflamatorias en una matriz mixoide.
- ✅ Variantes clínicas (OMS 2020):
- 🔹 Neurofibroma localizado (90%): Solitario, esporádico, superficial o profundo. Bajo riesgo de transformación.
- 🔹 Neurofibroma difuso: Infiltrativo, afecta piel y tejido subcutáneo, típico de niños y adultos jóvenes.
- 🔹 Neurofibroma plexiforme: Patognomónico de NF1 (30-50% de los pacientes). Afecta largos segmentos nerviosos, aspecto multinodular en «bolsa de gusanos». Riesgo de transformación a MPNST: 8-12%.
- ✅ Asociación sindrómica: Neurofibromatosis tipo 1 (NF1) por mutación en el gen NF1 (17q11.2), que codifica la neurofibromina. Criterios diagnósticos: ≥2 de: ≥6 manchas café con leche, ≥2 neurofibromas cutáneos o 1 plexiforme, efélides axilares/inguinales, glioma óptico, nódulos de Lisch, lesiones óseas características (displasia esfenoides, pseudoartrosis), antecedente familiar.
- ✅ Clínica: Masa blanda, renitente, signo del botón de campana (invaginación con la presión). El neurofibroma plexiforme puede causar deformidades, hipertrofia de extremidades, dolor neuropático.
- ✅ Imagen (RM): Signo de la diana en T2: centro hipointenso (tejido fibroso), anillo hiperintenso (estroma mixoide).
- ✅ Histopatología: Proliferación fusocelular en estroma mixoide, con mastocitos, sin cápsula, con infiltración del nervio.
- ✅ Inmunohistoquímica: S100+ (débil, parcheado), SOX10+, CD34+ (fibroblastos).
- ✅ Tratamiento: Observación en neurofibromas localizados asintomáticos. Cirugía en sintomáticos o sospecha de transformación. Selumetinib (inhibidor MEK) aprobado para neurofibromas plexiformes sintomáticos e irresecables en NF1 (reduce volumen tumoral en >70%).
1) Conceptos generales
1.1 Definición y origen celular
El neurofibroma es un tumor benigno de la vaina nerviosa periférica, no encapsulado, que se origina a partir de una célula precursora de la cresta neural con capacidad de diferenciación hacia células de Schwann, fibroblastos y pericitos. A diferencia del schwannoma, que está compuesto exclusivamente por células de Schwann, el neurofibroma contiene una mezcla heterogénea de:
- Células de Schwann: Constituyen el componente neoplásico principal, con pérdida bialélica de NF1.
- Fibroblastos: CD34+.
- Pericitos.
- Mastocitos.
- Células inflamatorias.
- Matriz extracelular mixoide y colágena.
1.2 Neurofibroma vs. schwannoma: diferencias clave
| Característica | Neurofibroma | Schwannoma |
|---|---|---|
| Cápsula | Ausente (infiltra el nervio) | Presente (epineuro/perineuro) |
| Relación con el nervio | Concéntrica (engloba fascículos) | Excéntrica (desplaza fascículos) |
| Composición celular | Mezcla: células de Schwann, fibroblastos, pericitos | Exclusivamente células de Schwann |
| Signo de la diana (RM T2) | Presente (centro hipointenso, anillo hiperintenso) | Invertido (centro isointenso, periferia hiperintensa) |
| S100 | + débil, parcheado | +++ difuso, nuclear |
| CD34 | Positivo en fibroblastos | Negativo |
| Neurofibromatosis | NF1 (plexiforme) | NF2, schwannomatosis |
| Transformación maligna | 8-12% en NF1 (plexiforme) | Excepcional |
2) Variantes clínicas de neurofibroma (OMS 2020)
2.1 Neurofibroma localizado
- Frecuencia: 90% de todos los neurofibromas.
- Presentación: Solitario, esporádico, puede ser superficial (cutáneo) o profundo (nervio periférico).
- Clínica: Nódulo cutáneo o subcutáneo, blando, a veces pediculado. Signo del botón de campana (se invagina con la presión). Indoloro.
- Localización: Cualquier localización, más frecuente en tronco, cabeza, cuello, extremidades.
- Asociación con NF1: Puede ser esporádico o formar parte de NF1 (múltiples).
- Riesgo de transformación: Muy bajo.
2.2 Neurofibroma difuso
- Presentación: Infiltrativo, afecta piel y tejido subcutáneo de forma difusa, sin formar nódulos definidos.
- Edad: Típico de niños y adultos jóvenes.
- Localización: Cabeza, cuello, tronco.
- Histología: Proliferación difusa de células fusiformes en estroma mixoide, con frecuentes corpúsculos táctiles (cuerpos de Wagner-Meissner-like).
- Asociación con NF1: Puede asociarse a NF1.
2.3 Neurofibroma plexiforme
- Definición: Tumor benigno que afecta a largos segmentos nerviosos, con aspecto multinodular y fusiforme, descrito clásicamente como «bolsa de gusanos».
- Frecuencia: Presente en el 30-50% de los pacientes con NF1.
- Localización: Troncos nerviosos mayores (plexo braquial, plexo lumbar, nervios de extremidades, cabeza y cuello).
- Clínica: Masa blanda, mal definida, que puede causar deformidades, hipertrofia de extremidades, dolor neuropático, compresión de estructuras vecinas.
- Riesgo de transformación maligna: 8-12% a lo largo de la vida. Es la principal lesión precursora de MPNST en NF1.
- Patognomónico de NF1: Su presencia confirma el diagnóstico de NF1, incluso en ausencia de otros criterios.
2.4 Otras variantes
- Neurofibroma atípico: Con atipia nuclear, hipercelularidad, pero sin necrosis ni mitosis atípicas. Riesgo incierto de progresión.
- Neurofibroma mixoide: Predominio de estroma mixoide.
- Neurofibroma pigmentado.
3) Neurofibromatosis tipo 1 (enfermedad de von Recklinghausen)
3.1 Genética
- Gen: NF1 (17q11.2), gen supresor tumoral.
- Proteína: Neurofibromina, que regula negativamente la vía RAS/MAPK (actúa como GAP de RAS).
- Herencia: Autosómica dominante, penetrancia completa a los 20 años. 50% de los casos son esporádicos (mutación de novo).
3.2 Criterios diagnósticos (NIH 1987, actualizados)
Se requiere la presencia de ≥2 de los siguientes criterios:
- ≥6 manchas café con leche (>5 mm en prepúberes, >15 mm en púberes).
- ≥2 neurofibromas cutáneos de cualquier tipo o ≥1 neurofibroma plexiforme.
- Efélides axilares o inguinales.
- Glioma óptico.
- ≥2 nódulos de Lisch (hamartomas del iris).
- Lesiones óseas características: displasia del esfenoides, adelgazamiento de la cortical de huesos largos (pseudoartrosis congénita de tibia).
- Antecedente familiar de NF1 en primer grado.
3.3 Riesgo de transformación maligna
Los pacientes con NF1 tienen un riesgo del 8-12% de desarrollar un MPNST a lo largo de la vida, generalmente sobre un neurofibroma plexiforme preexistente. El riesgo es mayor en adultos jóvenes.
4) Clínica
4.1 Neurofibroma localizado
- Masa: Nódulo cutáneo o subcutáneo, de consistencia blanda, renitente, a veces pediculado. Signo del botón de campana (se invagina con la presión).
- Crecimiento: Lento.
- Síntomas: Generalmente asintomático. Puede haber dolor o parestesias si comprime un nervio.
4.2 Neurofibroma plexiforme
- Masa: Mal definida, blanda, que puede extenderse a lo largo de un nervio y sus ramas. Aspecto de «bolsa de gusanos» a la palpación.
- Deformidades: Hipertrofia de la extremidad afectada, asimetría facial, escoliosis.
- Dolor neuropático: Frecuente.
- Síntomas compresivos: Compresión de vía aérea, vasos, médula espinal.
- Signos de transformación maligna: Crecimiento rápido, dolor intenso de nueva aparición, cambio de consistencia (más duro), déficit neurológico progresivo.
4.3 Exploración física
- Inspección de la piel: manchas café con leche, efélides, neurofibromas cutáneos.
- Palpación de masas: consistencia, tamaño, movilidad, signo del botón de campana.
- Exploración neurológica: déficit sensitivo/motor, signo de Tinel.
- Evaluación de deformidades óseas: escoliosis, pseudoartrosis de tibia.
- Examen oftalmológico: nódulos de Lisch (lámpara de hendidura).
5) Imagen
5.1 Radiografía simple
- Neurofibroma intraóseo: Lesión lítica, bien delimitada, con bordes escleróticos.
- Neurofibroma plexiforme: Puede producir erosión ósea por presión, escoliosis, displasia del esfenoides.
5.2 Ecografía
- Masa sólida, hipoecoica, bien delimitada, con refuerzo posterior variable.
- Relación con el nervio visible en cortes transversales (concéntrica).
5.3 Resonancia magnética (RM) - PRUEBA DE ELECCIÓN
- T1: Señal isointensa o hipointensa.
- T2/STIR: Signo de la diana característico: centro hipointenso (tejido fibroso) y anillo hiperintenso (estroma mixoide).
- T1 con contraste: Realce variable, a menudo periférico.
- Neurofibroma plexiforme: Múltiples lesiones fusiformes a lo largo del nervio, con el mismo patrón de señal.
- Hallazgos de sospecha de transformación maligna:
- Pérdida del signo de la diana.
- Necrosis intratumoral.
- Realce periférico heterogéneo.
- Masa de partes blandas infiltrativa.
- Restricción en difusión (ADC bajo).
5.4 PET-TC con FDG
- Indicación: Sospecha de transformación maligna en neurofibroma plexiforme.
- Hallazgo: Captación intensa y heterogénea (SUVmax >3-5) sugiere MPNST. Sensibilidad >90%, especificidad 70-80%.
- Valor de corte: SUVmax >3.5 es altamente sugestivo de malignidad.
6) Histopatología
6.1 Macroscopía
- Masa gris-blanca, firme, de aspecto fasciculado, sin cápsula, que se fusiona con el nervio de origen.
- En el neurofibroma plexiforme, múltiples nódulos fusiformes a lo largo del nervio, dando aspecto de «bolsa de gusanos».
6.2 Microscopía
- Proliferación de células fusiformes con núcleos ondulados, en un estroma mixoide rico en fibras de colágeno tipo I.
- Mastocitos: Abundantes, característicos.
- Ausencia de cápsula.
- Infiltración del nervio: Las células tumorales engloban los fascículos nerviosos.
- Cuerpos táctiles: En el neurofibroma difuso, pueden verse corpúsculos de Wagner-Meissner-like.
- Atipia: En el neurofibroma atípico, puede haber atipia nuclear, hipercelularidad, pero sin mitosis atípicas ni necrosis.
7) Inmunohistoquímica
| Marcador | Expresión | Comentario |
|---|---|---|
| S100 | + (débil, parcheado) | Menos intenso que en schwannoma. |
| SOX10 | + (nuclear) | |
| CD34 | + (en fibroblastos) | Patrón reticular característico. |
| Neurofilamentos | + (axones atrapados) | Confirma infiltración nerviosa. |
| EMA | Negativo | Las células perineurales están ausentes. |
| Desmina, actina | Negativo |
8) Genética molecular
8.1 Neurofibroma esporádico
- Pérdida bialélica de NF1 en las células de Schwann neoplásicas.
- Sin otras alteraciones recurrentes.
8.2 Neurofibroma en NF1
- Mutación germinal en NF1 + pérdida somática del segundo alelo (second hit) en las células de Schwann.
- El estroma (fibroblastos, pericitos) no tiene la mutación.
8.3 Transformación a MPNST
- Alteraciones adicionales en:
- CDKN2A/B (pérdida, 9p21).
- TP53 (mutación, 17p13).
- SUZ12 y EED (pérdida de función, alteraciones epigenéticas).
- EGFR (sobrexpresión).
9) Diagnóstico diferencial
9.1 Con otros tumores de la vaina nerviosa
| Entidad | Clave diferencial |
|---|---|
| Schwannoma | Encapsulado, excéntrico, S100 difuso, CD34-, cuerpos de Verocay. |
| MPNST (sarcoma de vaina nerviosa) | Crecimiento rápido, dolor, atipia, mitosis, necrosis, pérdida H3K27me3. |
| Perineurioma | EMA+, S100-, SOX10-. |
9.2 Otros tumores de partes blandas
- Tumor de células granulares: S100+, SOX10+, pero citoplasma granular eosinófilo.
- Fibroma de la vaina tendinosa.
- Fibromatosis.
- Melanoma: S100+, SOX10+, HMB45+, Melan A+.
10) Tratamiento
10.1 Observación
- Indicación: Neurofibromas localizados asintomáticos, sin crecimiento.
- Seguimiento: Clínico anual.
10.2 Cirugía
- Indicación: Neurofibromas sintomáticos (dolor, déficit neurológico, compresión, deformidad) o con sospecha de transformación maligna.
- Técnica:
- Neurofibroma localizado: Resección difícil por la infiltración del nervio. Si es posible, se intenta preservar los fascículos funcionales (neuromonitorización).
- Neurofibroma plexiforme: Resección compleja, a menudo incompleta, con alto riesgo de déficit neurológico. La cirugía se reserva para casos con síntomas graves o sospecha de malignidad.
- Riesgos: Déficit neurológico postoperatorio (frecuente), recidiva (si resección incompleta).
10.3 Terapia médica: inhibidores de MEK
- Selumetinib: Inhibidor de MEK1/2 (vía RAS/MAPK). Aprobado por EMA/FDA (2020-2021) para neurofibromas plexiformes sintomáticos e irresecables en niños ≥2 años con NF1.
- Dosis: 25 mg/m² dos veces al día, vía oral.
- Resultados: Reducción del volumen tumoral >20% en el 70% de los pacientes, mejoría del dolor y de la calidad de vida. Respuestas mantenidas a largo plazo.
- Efectos adversos: Cardiotoxicidad (disminución de la fracción de eyección, hipertensión), toxicidad oftalmológica (retinopatía, desprendimiento de retina), elevación de CK, toxicidad gastrointestinal.
- Seguimiento: Monitorización cardiológica y oftalmológica obligatoria.
10.4 Tratamiento del dolor
- Analgésicos convencionales (AINEs, opioides) según intensidad.
- Fármacos para dolor neuropático: gabapentina, pregabalina, antidepresivos tricíclicos.
11) Pronóstico y seguimiento
11.1 Neurofibroma localizado
- Pronóstico: Excelente. Recurrencia local baja tras resección completa.
- Seguimiento: Clínico. No requiere seguimiento oncológico específico.
11.2 Neurofibroma plexiforme en NF1
- Pronóstico: Variable. La mayoría son de crecimiento lento, pero el riesgo de transformación maligna (8-12%) requiere vigilancia estrecha.
- Seguimiento:
- Clínico: Evaluación periódica de síntomas, tamaño y consistencia de las masas.
- RM: Cada 1-2 años para evaluar crecimiento y detectar cambios sugestivos de malignidad.
- PET-TC: Ante sospecha de transformación (crecimiento rápido, dolor, cambio en RM).
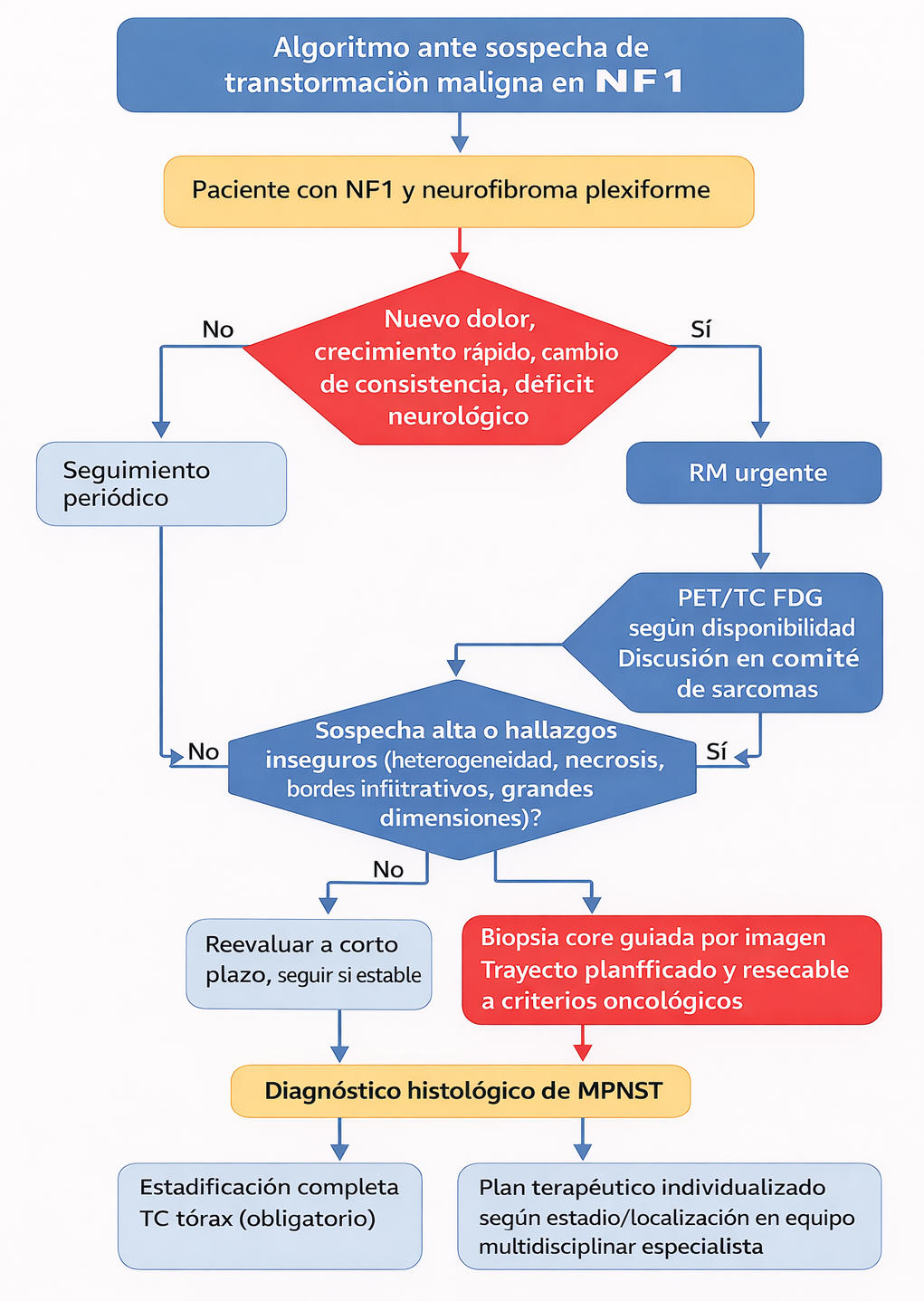
12) Algoritmo: cribado de transformación maligna en NF1
📌 Resumen para la práctica clínica
Indicaciones
- ✅ RM con neurografía en toda sospecha de neurofibroma plexiforme para caracterización y seguimiento.
- ✅ PET-TC con FDG ante sospecha de transformación maligna (crecimiento rápido, dolor, cambios en RM).
- ✅ Biopsia con aguja gruesa (core needle) en lesiones con sospecha de malignidad.
- ✅ Observación en neurofibromas localizados asintomáticos.
- ✅ Cirugía en neurofibromas sintomáticos (compresión, dolor) o con sospecha de transformación.
- ✅ Selumetinib en neurofibromas plexiformes sintomáticos e irresecables en NF1 pediátrica.
Técnica
- 🔧 RM: secuencias T1, T2/STIR, T1 con contraste y saturación grasa, DWI. Neurografía con tractografía.
- 🔧 PET-TC: ayunas 6h, glucemia <200 mg/dL, adquisición 60 min postinyección FDG.
- 🔧 Biopsia: aguja 14-16G, 3-5 pases, dirigida a áreas sólidas/nodulares (evitar zonas quísticas/necróticas).
Riesgos
- ⚠️ No sospechar transformación maligna en neurofibroma plexiforme con crecimiento rápido.
- ⚠️ Realizar cirugía sin neuromonitorización en neurofibromas que afectan nervios mayores.
- ⚠️ No ofrecer selumetinib a pacientes con NF1 y neurofibromas plexiformes sintomáticos.
- ⚠️ Confundir un neurofibroma con un schwannoma y realizar una cirugía inadecuada.
Resultados
- ✅ Neurofibroma localizado: recurrencia <5% tras resección completa.
- ✅ Neurofibroma plexiforme: reducción de volumen >20% en 70% de pacientes con selumetinib.
- ✅ MPNST: supervivencia global a 5 años 35-50%.
13) Bibliografía
📘 Clasificación y guías clínicas
- 📄 WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon: IARC Press; 2020. (Capítulo: Tumors of peripheral nerves).
- 📄 Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome. Genet Med. 2021;23(8):1506-1513. doi:10.1038/s41436-021-01170-8
🩻 Diagnóstico por imagen
- 📄 Ahlawat S, Blakeley JO, Langmead S, et al. Imaging of peripheral nerve sheath tumors: current status and future directions. Radiology. 2025;314(2):e240567. doi:10.1148/radiol.240567
- 📄 Broski SM, Johnson GB, Howe BM, et al. 18F-FDG PET/CT for detection of malignant transformation in neurofibromatosis type 1: a meta-analysis. Eur J Nucl Med Mol Imaging. 2024;51(4):1023-1035. doi:10.1007/s00259-023-06543-5
💊 Tratamiento con inhibidores de MEK
- 📄 Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430-1442. doi:10.1056/NEJMoa1912735
- 📄 Dombi E, Baldwin A, Marcus LJ, et al. Long-term safety and efficacy of selumetinib in pediatric NF1: 5-year follow-up. J Clin Oncol. 2025;43(2):210-220. doi:10.1200/JCO.24.01234
🧬 Genética y biología molecular
- 📄 Gutmann DH, Ferner RE, Listernick RH, et al. Neurofibromatosis type 1: update on pathogenesis and clinical management. Nat Rev Neurol. 2025;21(2):85-101. doi:10.1038/s41582-024-01052-1
🧭 Seguir navegando por este bloque
Has terminado esta ficha. Desde aquí puedes volver a la página troncal o pasar a otras entidades relacionadas.