Osteosarcoma — Clasificación y variantes — Actualización 2026: clasificación OMS 2020, características clínicas, radiológicas y patológicas
El osteosarcoma es el tumor óseo maligno primario más frecuente, caracterizado por la producción de matriz osteoide atípica por células tumorales malignas. Su clasificación actual (OMS 2020) distingue múltiples variantes con diferente comportamiento biológico, pronóstico y tratamiento. Esta ficha desarrolla de manera sistemática todas las variantes de osteosarcoma, desde el convencional central (80-85% de los casos) hasta las formas más raras como el osteosarcoma intracortical o el multicéntrico. Se incluyen para cada variante: epidemiología, clínica, hallazgos radiológicos, histopatología, inmunohistoquímica, genética molecular, pronóstico y opciones terapéuticas específicas. El objetivo es proporcionar una herramienta completa para el diagnóstico diferencial y el manejo de este complejo grupo de neoplasias.
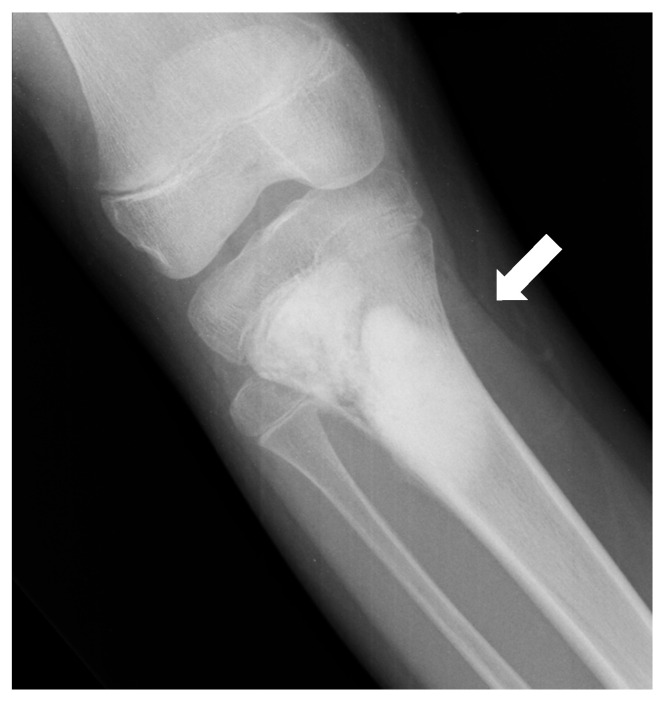
Hallazgo radiológico característico
Navegación rápida por variantes
Osteosarcoma central convencional (intramedular clásico)
Discusión general
💡 También conocido como osteosarcoma clásico, común o convencional. Es el tipo más frecuente, constituyendo el 80-85% de todos los osteosarcomas, aunque solo representa el 0.2% de todas las neoplasias malignas del hombre. Es el tumor maligno de hueso en el que la neoformación de osteoide o hueso se produce por células tumorales y constituye un criterio diagnóstico esencial.
Epidemiología
- 📈 Incidencia: Su incidencia más alta se da en la segunda década de la vida, coincidiendo con el estirón de crecimiento puberal.
- 📊 Sexo: Ligeramente más frecuente en hombres.
- 📏 Estatura: Se ha observado una incidencia superior en individuos de mayor estatura.
Localización
- 📍 Región de rodilla (50%): Fémur distal o tibia proximal.
- 📍 Otros sitios: Húmero proximal, fémur proximal, pelvis.
- 📍 Columna vertebral: Infrecuente.
- 📍 Localización en el hueso: La mayoría ocurre en la metáfisis. La placa de crecimiento supone una barrera relativa a la extensión tumoral, aunque se han descrito casos de invasión de la fisis, epífisis y articulación.
Presentación clínica
- 👤 Dolor: Síntoma inicial. Puede empezar insidiosamente, pero rápidamente se convierte en severo, constante y que no mejora con la inmovilización ni con analgésicos ordinarios. A menudo irradia a la articulación vecina.
- 👤 Tumoración palpable: Masa de partes blandas asociada.
- 👤 Limitación funcional: Impotencia funcional de la articulación vecina.
- 👤 Signos inflamatorios: En casos avanzados.
- 👤 Fractura patológica: Rara, más frecuente en lesiones predominantemente líticas.
- 👤 Fosfatasas alcalinas (FA): Suelen estar moderada o severamente elevadas, reflejando la actividad osteoblástica. Tienen valor pronóstico: descienden tras la cirugía y aumentan en recidivas o metástasis.
- 👤 Ganglios linfáticos: Afectación infrecuente.
- 👤 Metástasis pulmonares: Pueden estar presentes al diagnóstico en aproximadamente el 10% de los pacientes. Las micrometástasis indetectables están probablemente presentes en la mayoría de los pacientes al diagnóstico.
Estudios de imagen
- 🔹 Aspecto variable: Según la edad, localización, rapidez de crecimiento y producción de material óseo neoplásico.
- 🔹 Patrón de destrucción: Puede mostrar destrucción medular y cortical. Bordes generalmente borrosos, con amplia zona de transición. El tipo de destrucción puede ser moteado o permeativo, raramente geográfico.
- 🔹 Matriz tumoral: El grado de radiopacidad refleja la cantidad de producción ósea tumoral. Pueden aparecer como lesiones esclerosas puras, osteolíticas puras o, más frecuentemente, mixtas.
- 🔹 Reacción perióstica: Puede adoptar forma de "sol naciente" (sunburst), "púas de peine", triángulo de Codman y, menos frecuentemente, laminar en "hojas de cebolla".
- 🔹 Masa de partes blandas: Frecuente, con focos de esclerosis.
- 🔹 Extensión diafisaria: El tumor puede extenderse por el canal medular de la diáfisis, especialmente en húmero proximal.
- 🔹 Skip metastases: Nódulos aislados a lo largo de la diáfisis o que "saltan" a la articulación.
- 🔹 Importante para determinar la extensión en la médula ósea y la destrucción cortical.
- 🔹 TC torácico obligatorio para descartar metástasis pulmonares.
- 🔹 Igual o más efectiva que la TC para la determinación de la extensión intraósea y extraósea.
- 🔹 Demuestra el edema peritumoral.
- 🔹 Identifica componentes intratumorales: áreas celulares, necrosis, licuefacción.
- 🔹 Los componentes cartilaginosos son fácilmente distinguibles con gadolinio.
- 🔹 Muestra incremento en la captación del radiotrazador.
- 🔹 Puede mostrar focos metastásicos intraóseos o lesiones multicéntricas.
- 🔹 Raramente se usa en la actualidad.
- 🔹 Útil para mapear el tumor, ver el abastecimiento vascular e identificar áreas adecuadas para biopsia.
Histopatología
- 🔹 Aspecto parenquimatoso o encefaloide en zonas no osteógenas.
- 🔹 Zonas osteógenas más duras y menos vascularizadas.
- 🔹 El osteosarcoma se puede dividir en tres subtipos histológicos según el componente predominante: Osteoblástico, Condroblástico, Fibroblástico.
- 🔹 Gran pleomorfismo celular, con células grandes, fusiformes o poliédricas.
- 🔹 Polimorfismo de los nucleolos, atipias, hipercromía.
- 🔹 Mitosis frecuentes y atípicas.
- 🔹 Material intercelular: tejido osteoide, óseo, fibroso o cartilaginoso con distribución anárquica.
- 🔹 Áreas de necrosis y hemorragia.
Evolución y pronóstico
- 📊 Evolución: Rápida, excepto en tumores de tipo densificante.
- 📊 Metástasis: Principalmente por vía hematógena, a pulmones (80% a 1-2 años), esqueleto y otros lugares. Las metástasis linfáticas son raras.
- 📊 Estadio al diagnóstico: La mayoría están en estadio IIb (infiltración de tejido blando).
- 📊 Invasión de la fisis: En el 50% de pacientes adolescentes, el tumor penetra el platillo de crecimiento.
- 📊 Skip metastases: Presentes en el 25% de pacientes tras resección quirúrgica.
- 📊 Supervivencia: Con cirugía sola: 20%; con quimioterapia adyuvante: >50% a 5 años. Enfermedad metastásica al diagnóstico: peor pronóstico.
- 📊 Factores pronósticos: Respuesta a quimioterapia (necrosis >90% buena), márgenes quirúrgicos, tamaño tumoral, localización, presencia de metástasis.
Metástasis: aspectos prácticos
- ⚠️ Tiempo: Pueden ocurrir sincrónicamente (al diagnóstico) o metasincrónicamente (después).
- ⚠️ Supervivencia según sitio: Metástasis viscerales: muy pobre; óseas no operables: 0%; óseas operables: >10%; pulmonares operables: 30%.
- ⚠️ Tiempo de aparición y pronóstico: Sincrónicas: 20-40% desarrollan metástasis. Metasincrónicas tempranas: <10%. Metasincrónicas tardías: 30-50%.
- ⚠️ Diagnóstico de metástasis pulmonares: Un estudio mostró que de 51 casos con sospecha de metástasis pulmonares, solo el 57% demostraron tumor en la toracotomía; el resto eran falsos positivos (atelectasias, fibrosis). El porcentaje de confirmación aumenta con el número de nódulos: con 1 nódulo, solo el 31% eran metástasis; con >7 nódulos, el 100%.
- ⚠️ Tratamiento de metástasis: La cirugía agresiva de las metástasis (toracotomía) junto con quimioterapia se asocia a supervivencia a largo plazo en aproximadamente el 20% de los pacientes.
Tratamiento
- 🔹 Neoadyuvante (preoperatoria): Estándar actual. Objetivos: tratar micrometástasis, reducir el tumor, evaluar respuesta histológica.
- 🔹 Esquema MAP: Metotrexato a altas dosis (12 g/m²), Doxorrubicina, Cisplatino.
- 🔹 Metotrexato a altas dosis: Primer uso en 1948; Jaffe lo usó para sarcoma metastásico en 1972 (30% respuesta); Rosen lo introdujo como neoadyuvante en 1977 (50% respuesta). La respuesta histológica se correlaciona con los niveles séricos de metotrexato a las 24-48 horas. Existe gran variabilidad interindividual en los niveles séricos; la monitorización y el ajuste de dosis pueden ser beneficiosos.
- 🔹 Adyuvante (postoperatoria): Se completa según la respuesta histológica. Los pacientes con buena respuesta (>90% necrosis) continúan con el mismo esquema. Los de mala respuesta pueden cambiar a ifosfamida/etopósido (evidencia limitada).
- 🔹 Objetivo: Resección quirúrgica amplia con márgenes negativos (R0).
- 🔹 Salvamento del miembro vs. amputación: La amputación se asocia a menor recidiva local pero no a beneficio en supervivencia. No hay diferencias funcionales ni de calidad de vida demostradas consistentemente. La amputación es más cara a largo plazo por la necesidad de prótesis.
- 🔹 Opciones quirúrgicas para fémur distal: Amputación, reconstrucción endoprotésica, aloinjerto, compuesto aloinjerto/prótesis, osteogénesis de distracción (Ilizarov), artrodesis, rotaciónplastia.
- 🔹 Recidiva local: Más frecuente con resecciones marginales y con necrosis <90% tras quimioterapia.
Osteosarcoma de células pequeñas
Discusión
💡 Denominado históricamente "osteosarcoma primitivo multipotencial primario del hueso" (Hutter, 1966), "polihistioma" (Jacobson, 1977), "osteosarcoma de células pequeñas" (Sim y col., 1979) u "osteosarcoma mesenquimal" (Schajowicz, 1983). Supone el 1% de todos los osteosarcomas.
Epidemiología y localización
- 📍 Edad: Segunda década.
- 📍 Localización preferida: Fémur distal, húmero proximal, tibia proximal.
Radiografía
- 📸 Generalmente se presenta como una lesión radiolucente con bordes permeativos, asociada a una gran masa de tejidos blandos.
- 📸 Esta presentación recuerda a los tumores de células redondas, como el sarcoma de Ewing.
- 📸 En algunos casos puede presentarse como lesión esclerosa similar al osteosarcoma clásico.
- 📸 Importante reacción perióstica en la mayoría de los casos.
Histología
- 🧫 Acúmulos de células redondas pequeñas, bandas separadas de colágeno y fina matriz eosinófila que recuerda al sarcoma de Ewing.
- 🧫 Puede mostrar tinción positiva de glucógeno, como en el sarcoma de Ewing.
- 🧫 Claves diferenciales con sarcoma de Ewing:
- 🔹 Presencia de núcleos ovales y células fusiformes en algunas áreas.
- 🔹 Producción de osteoide o hueso (criterio diagnóstico esencial).
- 🔹 En algunos casos puede haber cartílago maligno.
- 🧫 Se han descrito translocaciones 11/22 en osteosarcomas de células pequeñas, lo que plantea la cuestión de si deben tratarse como osteosarcoma o como sarcoma de Ewing.
Osteosarcoma fibrohistiocítico
Discusión
💡 Esta variante recuerda al fibrohistiocitoma maligno (actualmente sarcoma pleomórfico indiferenciado). Pueden confundirse, ya que ambos tumores tienden a aparecer a una edad superior a la del osteosarcoma clásico (generalmente después de la tercera década) y afectan a la porción articular de los huesos largos. La reacción perióstica es menor que en el osteosarcoma convencional.
Radiografía
- 📸 Ambas lesiones suelen ser radiolucentes.
- 📸 El osteosarcoma fibrohistiocítico presenta áreas de formación ósea que asemejan "bolas de algodón" o "acúmulos de nubes", mientras que el fibrohistiocitoma maligno no tiene esta apariencia.
Histología
- 🧫 Presencia de células fusiformes pleomórficas y células gigantes, muchas con núcleos extravagantes. Recuerda al osteosarcoma rico en células gigantes.
- 🧫 No es normal un fondo inflamatorio.
- 🧫 La configuración nebulosa estoriforme o espiral, característica del fibrohistiocitoma, puede ser un hallazgo dominante, menos prominente o estar sustituida por grandes áreas de células pleomórficas en un manto difuso.
- 🧫 Clave diagnóstica: Demostración de formación de osteoide o hueso por las células malignas en el típico patrón de osteosarcoma.
Osteosarcoma telangiectásico
Discusión
💡 También llamado osteosarcoma hemorrágico. Es muy raro (5% de los osteosarcomas) y muy agresivo. Es el doble de frecuente en mujeres que en hombres, y predomina en la segunda y tercera década. Suele aparecer en fémur o tibia. Es un tumor hipervascularizado con zonas necróticas y hemorrágicas, que presenta grandes sinusoides, cavidades hemáticas y células gigantes multinucleadas reaccionales a la hemorragia.
Radiografía
- 📸 Se presenta como una lesión lítica y destructiva exclusivamente.
- 📸 Puede haber masa de tejidos blandos.
- 📸 Reacción perióstica agresiva: laminar, en sol naciente, triángulo de Codman.
- 📸 En ocasiones recuerda al quiste óseo aneurismático.
RM
- 💡 Muestra áreas de alta señal en T1 debidas a la presencia de hemoglobina.
- 💡 Pueden verse niveles líquido-líquido, indicando la presencia de hemorragias (también presentes en quiste óseo aneurismático).
Macroscopía
- 🔍 Tumor muy blando, semejando a una esponja con grandes cavidades llenas de sangre y coágulos.
- 🔍 Gran destrucción de la cortical y masa de partes blandas.
Histología
- 🔬 Grandes cavidades entremezcladas con tabiques y zonas con origen sarcomatoso.
- 🔬 A poco aumento recuerda a un quiste óseo aneurismático.
- 🔬 A mayor aumento: elementos de origen mesenquimatoso con gran pleomorfismo, muchas mitosis y células gigantes benignas y malignas.
- 🔬 Producción focal de osteoide (mucho mayor que en el quiste óseo aneurismático). En ocasiones, la producción de osteoide es mínima.
Evolución y pronóstico
- ⚠️ Evolución rápida y agresiva.
- ⚠️ Aunque responde bien a la quimioterapia, su agresividad puede no dar tiempo a completar los ciclos de quimioterapia preoperatoria, teniendo que recurrir directamente a la amputación.
Osteosarcoma rico en células gigantes
Discusión
💡 Variedad rara de osteosarcoma (3% de todos los osteosarcomas), con apariencia de sarcoma indiferenciado con hiperabundancia de osteoclastos (células gigantes) y escasez de osteoide o hueso tumoral. Está relacionado histológicamente con el osteosarcoma telangiectásico y con el fibrohistiocítico.
Radiografía
- 📸 Ausencia de muchos de los signos clásicos del osteosarcoma.
- 📸 Lesión lítica con bordes pobremente definidos.
- 📸 Localización típica: metáfisis o diáfisis de huesos largos (tibia, fémur).
- 📸 Reacción perióstica ausente o escasa.
- 📸 Generalmente no hay masa de tejidos blandos.
Histología
- 🧫 Estrecho parecido al tumor de células gigantes.
- 🧫 Producción de osteoide escasa y difícil de identificar, clave diagnóstica.
Osteosarcoma central de bajo grado (bien diferenciado)
Discusión
💡 Variedad muy rara (<2% de todos los osteosarcomas). Generalmente afecta a pacientes mayores que los del osteosarcoma clásico (media 28 años). Sus sitios predilectos son la tibia y el fémur. Es un tumor central, osteolítico, condensante o mixto, que presenta un patrón radiológico poco agresivo.
Clínica
- 👤 Larga historia de dolor, disconfort o tumefacción inespecífica.
Radiografía
- 📸 En muchos casos se etiqueta de displasia fibrosa en base a la radiografía y a la histología.
- 📸 En otros casos es indistinguible del osteosarcoma central clásico.
- 📸 Más a menudo, la lesión aparenta benignidad, con anillo escleroso bien definido.
- 📸 Expansión frecuente, cortical adelgazada.
- 📸 Reacción perióstica rara, así como extensión a tejidos blandos.
- 📸 El diagnóstico diferencial radiológico incluye: displasia fibrosa, tumor de células gigantes, fibroma desmoplástico, ocasionalmente miositis osificante.
Histología
- 🧫 Diagnóstico histológico muy difícil. Puede parecerse a displasia fibrosa, fibroma no osificante, osteoblastoma, fibroma condromixoide o reacciones inespecíficas.
- 🧫 Campos fibroblásticos con trabéculas óseas y osteoides.
- 🧫 Las células intercaladas entre las trabéculas son fusiformes, organizadas en manojos, sin apariencia de malignidad.
- 🧫 El núcleo puede mostrar alguna atipia, pero no hay pleomorfismo y las mitosis son raras.
- 🧫 Puede haber producción de colágeno y hueso.
- 🧫 Las células estromales pueden invadir la cavidad medular y las trabéculas del hueso esponjoso y cortical.
- 🧫 La lesión puede parecerse al fibroma desmoplástico en unos casos y a la displasia fibrosa en otros. Se diferencian de esta última porque las espículas en la displasia fibrosa son generalmente voluminosas, poco delicadas y de forma curvilínea.
- 🧫 Pueden verse focos de cartílago (raro).
- 🧫 El diagnóstico solo es posible con la ayuda de la correlación radiográfica.
Evolución, pronóstico y tratamiento
- 💡 Evolución: Lenta o muy lenta.
- 💡 Pronóstico: Bueno, aunque se han descrito casos con metástasis.
- 💡 Tratamiento de elección: Resección quirúrgica amplia, sin necesidad de quimioterapia.
Osteosarcoma intracortical
Discusión
💡 Es la forma más rara de osteosarcoma (solo 8 casos comunicados). El tumor afecta la cortical y no se extiende a la médula ni al tejido blando. El rango de edad oscila entre 10 y 40 años (media 24), con predominio femenino.
Clínica
- 👤 Dolor, frecuentemente asociado con la actividad.
- 👤 En algunos pacientes hay historia de traumatismo previo.
Radiografía
- 📸 Lesión radiolucente rodeada de una reacción esclerosa.
- 📸 Tamaño variable de 1 a 4.2 cm.
- 📸 En algunos casos puede evocar a un osteoma osteoide o un osteoblastoma intracortical.
Histología
- 🧫 Células tumorales redondas o fusiformes, pleomórficas, con núcleo vesicular y prominentes nucleolos.
- 🧫 Figuras mitóticas.
- 🧫 La matriz producida por las células malignas tiene un patrón acordonado con osteoide tumoral.
Osteosarcoma maxilar
- 💡 Se da en pacientes de más edad que el osteosarcoma convencional (35 años de media).
- 💡 Generalmente es un tumor bien diferenciado, con índice bajo de mitosis, un gran componente condroblástico, y un pobre potencial de malignidad.
- 💡 Su pronóstico es mejor que otras formas de osteosarcoma, aunque su localización impide la cirugía amplia.
Osteosarcoma multicéntrico (multifocal)
Discusión
💡 También llamado osteosarcomatosis esclerosante. Se trata de focos múltiples de osteosarcomas esclerosantes que aparecen, al mismo tiempo (sincrónicos) o en un intervalo corto (metacrónicos), en diferentes lugares del esqueleto. Es un tipo muy raro (1.5% de todos los osteosarcomas).
Epidemiología
- 📈 Edad: Suele presentarse antes de los 18 años.
- 📈 Sexo: Ligero predominio femenino en edades superiores.
- 📈 Distribución: Muy difusa. Los focos pueden ser muy numerosos y algunos muy pequeños. Sitios preferidos: tercio distal del fémur, proximal de la tibia, proximal del fémur, pelvis, húmero proximal.
Clasificación de Amstutz
- 📌 Tipo I (sincrónico): Pacientes jóvenes, sin metástasis pulmonares en la mayoría, lesiones densas y esclerosas en metáfisis de huesos largos. Histología: densa osificación osteoblástica.
- 📌 Tipo II: Lesiones de bajo grado, sincrónicas, limitadas al esqueleto axial. Adultos.
- 📌 Tipo III (metacrónico): Lesiones esqueléticas asimétricas y de tamaño variable, no necesariamente esclerosas. Edad variable. En algunos casos se cree que se trata de metástasis de un osteosarcoma primario.
Controversia sobre la naturaleza
🤔 No está claro si este tipo de osteosarcoma es una entidad separada o se trata de múltiples metástasis de un osteosarcoma convencional. Cuando hay múltiples huesos afectados en ausencia de metástasis pulmonares, lo más probable es que se trate de un osteosarcoma multicéntrico verdadero.
Características radiológicas y patológicas
- 📸 Los focos de osteosarcoma tienen la peculiaridad de ser uniformemente condensantes o ebúrneos y de no atravesar la cortical.
Pronóstico
- ⚠️ Osteosarcoma multifocal sincrónico: Pronóstico muy pobre. La mayoría de los pacientes sobreviven unos pocos meses después del diagnóstico.
- ⚠️ Lesiones metacrónicas: Potencialmente curables con tratamiento quirúrgico.
Bibliografía
Referencias clave
- 📄 WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon: IARC Press; 2020.
- 📄 Gronchi A, Miah AB, Dei Tos AP, et al. Soft tissue and visceral sarcomas: ESMO-EURACON-GENTURIS Clinical Practice Guidelines. Ann Oncol. 2024;35(8):678-695.
- 📄 Marina NM, Gorlick R, Bielack SS, et al. EURAMOS-1: 15-year follow-up of methotrexate, doxorubicin, and cisplatin in osteosarcoma. Lancet Oncol. 2025;26(4):512-524.
Resumen para la práctica clínica
📌 Indicaciones
- ✅ Radiografía simple en dos proyecciones (prueba inicial fundamental).
- ✅ RM con contraste para evaluar extensión intraósea y de partes blandas.
- ✅ TC torácico para descartar metástasis pulmonares (obligatorio).
- ✅ Biopsia con aguja gruesa (core needle) con material suficiente para IHC y genética.
- ✅ Determinación de fosfatasas alcalinas (FA) y LDH séricas.
🔧 Técnica
- 🔧 RM: secuencias T1, T2/STIR, T1 con contraste. Campo de visión amplio incluyendo todo el hueso.
- 🔧 TC torácico: cortes finos con reconstrucción de alta resolución.
- 🔧 Biopsia: guiada por imagen, trayecto planificado con el cirujano, evitar zonas necróticas.
- 🔧 IHC: SATB2, osteocalcina, vimentina, MDM2 (en bajo grado).
⚠️ Riesgos
- ⚠️ Realizar biopsia sin un estudio de imagen completo previo.
- ⚠️ Confundir osteosarcoma de bajo grado con displasia fibrosa (error diagnóstico).
- ⚠️ No sospechar osteosarcoma telangiectásico en una lesión lítica con niveles líquido-líquido.
- ⚠️ Asumir que un osteosarcoma multicéntrico es una enfermedad metastásica incurable sin valorar la posibilidad de lesiones metacrónicas operables.
✅ Resultados
- ✅ Osteosarcoma convencional localizado: SG 70-80% con tratamiento actual.
- ✅ Osteosarcoma de bajo grado: excelente pronóstico con cirugía sola.
- ✅ Osteosarcoma telangiectásico: pronóstico similar al convencional con quimioterapia.