Sarcomas de partes blandas — subpágina oncológica (Actualización 2026): estadificación, pronóstico y tratamiento especializado tras la sospecha inicial
Los sarcomas de partes blandas (SPB) son tumores malignos raros de origen mesenquimal (músculo, grasa, tejido fibroso, vasos, nervios), con una incidencia global en Europa alrededor de 4–5 casos/100.000/año. Esta página está pensada como subpágina hija de tumores de partes blandas: parte de que la sospecha inicial, la imagen básica y los principios generales de biopsia ya se han entendido en la troncal, y se centra en lo específicamente oncológico: estadificación, pronóstico, cirugía R0, radioterapia perioperatoria, tratamiento sistémico y manejo en comité de sarcomas. La actualización 2026 refuerza cuatro ideas: derivación temprana a unidades de sarcoma, biopsia planificada (evitar “exéresis no planificada”), revisión anatomopatológica experta con IHQ + biología molecular/NGS cuando aporte valor diagnóstico o terapéutico, y un enfoque multimodal centrado en cirugía R0 + radioterapia perioperatoria en tumores de riesgo, reservando tratamiento sistémico para subtipos sensibles, alto riesgo seleccionado o enfermedad avanzada.
← Volver a la página troncal de tumores de partes blandas · Ver simuladores de sarcoma y pitfalls diagnósticos
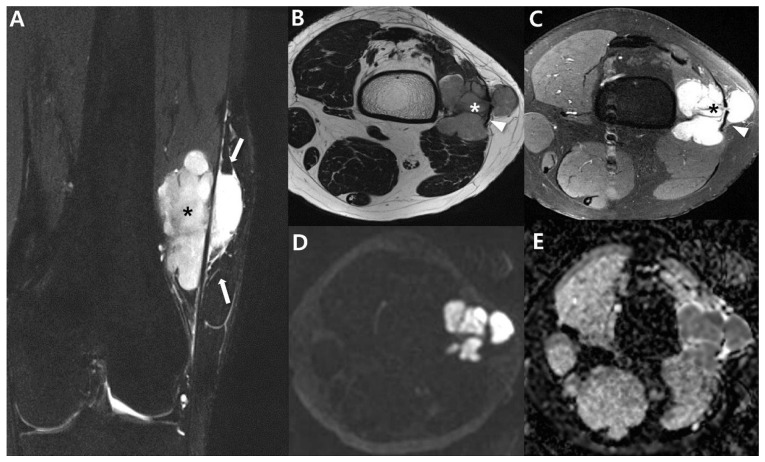
Hallazgo radiológico característico
0) En 1 minuto
- ✅ Papel de esta página: desarrollar el bloque maligno de la troncal de partes blandas; no sustituye la orientación general ante toda masa de partes blandas.
- ✅ Cuándo entrar en esta ficha: cuando una masa ya es sospechosa de sarcoma por red flags, imagen o biopsia, o cuando necesitas el plan oncológico del bloque maligno.
- ✅ Imagen clave: RM con contraste para estadificación local (extensión, fascias, vasos/nervios) y TC de tórax para metástasis pulmonares en sarcomas de riesgo.
- ✅ Diagnóstico: biopsia core-needle (o incisional si precisa), siempre planificada por el equipo que tratará. Anatomía patológica experta con IHQ y pruebas moleculares/NGS cuando confirmen entidad o abran terapias dirigidas.
- ✅ Tratamiento localizado: cirugía R0 (resección en bloque) + radioterapia perioperatoria en tumores G2–G3, ≥5 cm, profundos o con márgenes comprometidos. Quimioterapia perioperatoria: seleccionada (alto riesgo/subtipo sensible).
- ✅ Enfermedad avanzada: base con antraciclinas (p. ej., doxorrubicina) y opciones por subtipo; existen terapias dirigidas (p. ej., NTRK-fusión) e inmunoterapia en subtipos concretos.
Pitfall: el error que más empeora resultados es la exéresis no planificada (“quitar el bulto” sin RM/biopsia ni márgenes oncológicos). Aumenta recidiva local, complica la resección definitiva y puede obligar a cirugías más mutilantes o a más radioterapia. En SPB: primero RM + biopsia bien hecha, y después tratamiento en comité multidisciplinar.
1) Conceptos
1.1 Qué incluye “sarcoma de partes blandas”
“SPB” engloba neoplasias malignas mesenquimales extraesqueléticas. Pueden aparecer en cualquier localización; las más frecuentes son extremidades (muslo), tronco y retroperitoneo. Su heterogeneidad es clínica y biológica: hay tumores impulsados por fusiones génicas (p. ej., SS18-SSX, EWSR1-ETS, FUS-DDIT3) y tumores con genómica compleja (p. ej., UPS), lo que condiciona diagnóstico y, a veces, tratamiento.
1.2 Dónde se decide el tratamiento: unidad de sarcomas
- ✅ Comité multidisciplinar (cirugía oncológica/trauma, radiología, anatomía patológica, oncología médica, oncología radioterápica, rehabilitación).
- ✅ Revisión central de anatomía patológica cuando el subtipo o el grado condicionan el plan.
- ✅ Planificación de biopsia y de la resección definitiva desde el inicio (trayecto excindible).
2) Epidemiología
- 📈 Incidencia (Europa): alrededor de 4–5/100.000/año (adultos; excluyendo GIST).
- 📈 Edad: más frecuente en adultos (pico en edades medias-avanzadas), aunque algunos subtipos predominan en niños/AYA (p. ej., rabdomiosarcoma, sarcoma sinovial).
- 📈 Localización más habitual: extremidades (muslo), seguida de tronco y retroperitoneo (según series).
- 📈 Metástasis inicial: variable según grado/subtipo; el pulmón es el destino más frecuente en sarcomas de alto grado.
3) Etiología
3.1 Predisposición genética
- 🔹 NF1: aumenta el riesgo de MPNST.
- 🔹 Li-Fraumeni (TP53): riesgo elevado de sarcomas (y otros tumores).
- 🔹 RB1 (retinoblastoma hereditario): mayor riesgo de sarcomas, especialmente tras radioterapia.
- 🔹 Otros síndromes raros: predisposición variable; valorar consejo genético si historia familiar/edad inusual/múltiples tumores.
3.2 Factores adquiridos
- 🔹 Radiación previa: sarcomas radioinducidos tras latencia (típicamente años).
- 🔹 Linfedema crónico: riesgo de angiosarcoma (Stewart–Treves).
- 🔹 Exposiciones específicas: p. ej., cloruro de vinilo (angiosarcoma hepático) en contextos concretos.
- 🔹 Trauma: no se considera causa; puede facilitar el hallazgo de una lesión preexistente.
4) Clasificación (orientativa, basada en OMS 5ª ed.)
La clasificación OMS se organiza por línea de diferenciación y por entidades definidas por genética. La tabla siguiente es práctica (no exhaustiva): agrupa familias y ejemplos frecuentes.
| Familia | Ejemplos benignos/intermedios | Ejemplos malignos (sarcomas) |
|---|---|---|
| Adipocíticos | Lipoma, lipomatosis | Liposarcoma (bien diferenciado/desdiferenciado, mixoide, pleomórfico) |
| Fibroblásticos / miofibroblásticos | Fibromatosis (desmoide), nodular fasciitis (reactiva) | Fibrosarcoma, SEF/LGFMS (MUC4+), sarcoma pleomórfico indiferenciado (UPS) |
| Músculo liso | Leiomioma | Leiomiosarcoma |
| Músculo esquelético | Rabdomioma (raro) | Rabdomiosarcoma (embrionario, alveolar, pleomórfico) |
| Vasculares | Hemangioma, malformaciones vasculares | Angiosarcoma, sarcoma de Kaposi (contextos específicos) |
| Nervio periférico | Schwannoma, neurofibroma | MPNST (incluye “tumor Tritón” como variante) |
| De diferenciación incierta / definidos por fusión | — | Sarcoma sinovial (SS18-SSX), sarcomas de células redondas (subgrupos), sarcoma de células claras |
| Varios / específicos | Tumor fibroso solitario (SFT; espectro) | SFT maligno, sarcoma epitelioide (INI1/SMARCB1), ASPS |
Nota: “GIST” es un tumor mesenquimal digestivo con guías y tratamiento propios; suele excluirse de SPB “adult-type” en muchas guías.
5) Clínica y criterios de derivación
- 🔹 Presentación típica: masa de crecimiento progresivo, muchas veces indolora, sobre todo si es profunda.
- 🔹 Red flags (derivar a unidad de sarcomas): >5 cm, profunda, creciente, dolor, recidiva tras cirugía previa, fijación a planos profundos, síntomas compresivos.
- 🔹 Ganglios: poco frecuente en SPB en general, pero más probable en subtipos concretos (p. ej., sarcoma epitelioide, células claras, rabdomiosarcoma, angiosarcoma, sinovial en algunas series).
| Dato | Orientación práctica |
|---|---|
| Masa superficial pequeña y estable | frecuente que sea benigna, pero si crece o es atípica → imagen y valoración. |
| Masa profunda o >5 cm | tratar como “sarcoma hasta demostrar lo contrario”: RM + biopsia planificada. |
| Dolor | no descarta benignidad; en sarcoma puede ser tardío (compresión/invasión). |
| Recidiva post-exéresis | alto riesgo de “exéresis no planificada”: reevaluación completa y resección ampliada. |
6) Evaluación diagnóstica
6.1 Imagen (antes de pinchar)
- 🔹 Ecografía: útil como primer filtro en masas superficiales; si es profunda/atípica → RM.
- 🔹 RM con contraste: técnica de elección para estadificación local (tamaño real, compartimentos, fascias, vasos/nervios, “skip lesions” en trayectos).
- 🔹 TC: útil en retroperitoneo/visceral, y para valorar calcificación/afectación ósea.
- 🔹 TC de tórax: recomendada en tumores de riesgo (alto grado/tamaño) para descartar metástasis pulmonares.
- 🔹 PET-TC: no es rutinario; puede ayudar en casos seleccionados (alto grado, respuesta a tratamiento, dudas de extensión).
6.2 Biopsia (principios “2026”)
- 🔹 Preferente: core-needle guiada (US/TC) con múltiples cilindros, orientada a zonas sólidas viables.
- 🔹 Incisional: si la core no es diagnóstica o se requiere más material (IHQ amplia, molecular).
- 🔹 Escisional: solo si lesión muy pequeña y superficial y con sospecha baja (excepción, no norma).
- 🔹 Regla de oro: la biopsia la planifica el mismo equipo que hará la cirugía definitiva; el trayecto debe ser excindible.
- 🔹 Evitar: hematomas (hemostasia), drenajes innecesarios y abordajes transversales en extremidades.
6.3 Anatomía patológica: grado, IHQ y genética
- 🔹 Grado: usar un sistema reproducible (p. ej., FNCLCC) porque condiciona estadificación y tratamiento.
- 🔹 IHQ orientativa:
- 🔹 MDM2/CDK4: liposarcoma bien diferenciado/desdiferenciado.
- 🔹 STAT6: tumor fibroso solitario.
- 🔹 S100/SOX10: tumores de vaina nerviosa / melanoma (diferencial).
- 🔹 Desmina/actina/caldesmón: músculo liso; miogenina/MyoD1 en rabdomiosarcoma.
- 🔹 INI1/SMARCB1: útil en epitelioide/rabdoide (pérdida).
- 🔹 MUC4: sugiere LGFMS/SEF (no “sinovial”).
- 🔹 Marcadores epiteliales (CK/EMA) + TLE1/SS18-SSX: apoyo en sarcoma sinovial (según contexto).
- 🔹 Genética/molecular: FISH/RT-PCR o NGS cuando:
- 🔹 confirma una entidad (p. ej., fusiones específicas),
- 🔹 cambia el manejo (p. ej., NTRK-fusión),
- 🔹 o resuelve un diferencial crítico.
7) Estadificación (AJCC 8ª edición: extremidad/tronco)
La AJCC 8ª para SPB de extremidades y tronco se basa en T (tamaño), N, M y grado. Importante: existen esquemas específicos para retroperitoneo, cabeza y cuello y otras localizaciones. El grado suele establecerse con FNCLCC.
- 🔹 T: T1 ≤5 cm; T2 >5–10; T3 >10–15; T4 >15 cm.
- 🔹 N: N1 (afectación ganglionar) es infrecuente pero, cuando existe, implica estadio avanzado.
- 🔹 M: M1 (metástasis a distancia).
- 🔹 G: G1–G3 (o GX si no evaluable).
| Estadio | Grado | T | N/M |
|---|---|---|---|
| I | G1 (o GX) | Cualquier T | N0 M0 |
| II | G2–G3 | T1 (≤5 cm) | N0 M0 |
| III | G2–G3 | T2–T4 | N0 M0 |
| IV | Cualquier G | Cualquier T | N1 o M1 |
En retroperitoneo, la estrategia y el pronóstico dependen mucho de histotipo (p. ej., liposarcoma vs leiomiosarcoma) y resecabilidad; la estadificación y el plan suelen diferir.
8) Pronóstico (lo que realmente manda)
- 📈 Grado (FNCLCC): principal predictor de metástasis y mortalidad.
- 📈 Tamaño: a mayor tamaño, mayor riesgo (y suele correlacionar con profundidad/compartimentos).
- 📈 Localización: retroperitoneo y algunas áreas anatómicas complejas tienen peor control local por dificultad de márgenes.
- 📈 Márgenes: R0 reduce recidiva local; R1/R2 empeoran control local y pueden impactar supervivencia según histotipo.
- 📈 Histotipo: algunos tienen patrón metastásico distinto y sensibilidad diferente a sistémicos.
- 📈 Biología: genética (fusiones, amplificaciones, alteraciones accionables) y marcadores de proliferación pueden refinar riesgo, pero su uso depende del subtipo.
Práctico: en la vida real, “grado + tamaño + localización + márgenes” decide casi todo el plan local; el “apellido” histológico decide matices y el tratamiento sistémico.
9) Tratamiento
Premisa: tratamiento en centros de referencia. Objetivos: control local durable y preservación funcional, minimizando toxicidad.
9.1 Cirugía (pilar del tratamiento localizado)
- 🔹 Objetivo: resección en bloque con margen negativo (R0). En extremidad, márgenes “funcionales” pueden ser aceptables si se combina con RT (según riesgo y planos barrera).
- 🔹 Evitar: resecciones marginales “a ciegas” y reintervenciones improvisadas.
- 🔹 Retroperitoneo: suele requerir resección multivisceral “en bloque” en manos expertas.
9.2 Radioterapia perioperatoria (control local en tumores de riesgo)
- 🔹 Indicaciones típicas: tumores G2–G3, ≥5 cm, profundos, cercanos a estructuras críticas o con márgenes cercanos/positivos.
- 🔹 Preoperatoria: suele usar ≈50 Gy en fracciones convencionales; menor volumen irradiado, pero más complicaciones de herida.
- 🔹 Postoperatoria: habitual si no se hizo preoperatoria y el riesgo local lo justifica; dosis típicas ≈60–66 Gy según escenario.
- 🔹 Técnicas: IMRT/VMAT; protonterapia en casos seleccionados para reducir dosis a órganos críticos.
9.3 Tratamiento sistémico (cuándo sí)
- 🔹 Perioperatorio: no es “de rutina” en todos los SPB; considerar en alto riesgo seleccionado y/o subtipos más quimiosensibles, idealmente en protocolo o tras discusión en comité.
- 🔹 Avanzado/metastásico:
- 🔹 Base general: doxorrubicina (± ifosfamida si se busca mayor respuesta).
- 🔹 Opciones por histotipo (ejemplos): gemcitabina-docetaxel (leiomiosarcoma), trabectedina (lipo/LMS), eribulina (lipo), pazopanib (muchos no adipocíticos), taxanos (angiosarcoma), etc.
- 🔹 Dirigidas “agnósticas”: tumores con NTRK-fusión (inhibidores TRK).
- 🔹 Inmunoterapia en subtipos concretos: p. ej., ASPS no resecable/metastásico (según disponibilidad/regulación).
- 🔹 Otras dirigidas por subtipo: p. ej., tazemetostat en sarcoma epitelioide avanzado no resecable (según indicación y acceso).
| Escenario | Estrategia |
|---|---|
| Localizado bajo riesgo | Cirugía R0; RT solo si riesgo local (márgenes, localización, recaída). |
| Localizado alto riesgo | RT preop o postop + cirugía R0; valorar sistémico perioperatorio en seleccionados/subtipos sensibles. |
| Irresecable | Convertir a resecable si posible (RT ± sistémico); si no, control local/funcional y sistémico según subtipo. |
| Metastásico | Sistémico por histotipo; considerar cirugía/RT de metástasis en oligometástasis seleccionada y control del primario. |
10) Algoritmo de manejo

Resumen para la práctica clínica
📌 Indicaciones
- ✅ Derivación a unidad de sarcomas ante masa >5 cm, profunda, creciente o recidivada.
- ✅ Resección quirúrgica en bloque con márgenes negativos (R0) en sarcomas localizados resecables.
- ✅ Radioterapia perioperatoria en tumores de alto riesgo (G2–G3, ≥5 cm, profundos o con riesgo de márgenes comprometidos).
- ✅ Tratamiento sistémico según histotipo/escenario: avanzado/metastásico y casos seleccionados de alto riesgo o subtipos sensibles.
🔧 Técnica
- 🔧 Imagen antes de biopsia: RM con contraste (o TC según localización).
- 🔧 Biopsia: core-needle guiada preferible, trayecto excindible, planificada por el equipo que realizará la cirugía definitiva.
- 🔧 Radioterapia: técnicas conformadas (IMRT/VMAT; protones en seleccionados). Preop ~50 Gy; postop ~60–66 Gy según escenario.
- 🔧 Patología: grado (p. ej., FNCLCC) + IHQ; biología molecular/NGS cuando confirme entidad o aporte opciones terapéuticas.
⚠️ Riesgos
- ⚠️ Exéresis no planificada con contaminación de márgenes y aumento de recidiva local.
- ⚠️ Recidiva local (riesgo ligado a márgenes, grado, tamaño y localización).
- ⚠️ Metástasis (especialmente pulmón en alto grado).
- ⚠️ Complicaciones de herida (más frecuentes con RT preoperatoria).
- ⚠️ Toxicidad de RT (fibrosis/edema) y de sistémicos (p. ej., cardiotoxicidad por antraciclinas).
✅ Resultados
- ✅ Pronóstico muy variable según grado, tamaño, localización, márgenes e histotipo; individualizar riesgo y seguimiento en unidad experta.
11) Bibliografía
Guías y clasificación
- 📄 WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. IARC; 2020.
- 📄 Gronchi A, Miah AB, Dei Tos AP, et al. Soft tissue and visceral sarcomas: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines. Ann Oncol. 2021;32(11):1348-1365.
- 📄 Serrano C, Arregui M, et al. SEOM-GEIS Spanish clinical guidelines for the management of soft-tissue sarcomas (2024). Clin Transl Oncol. 2025;27(4):1460-1471. doi:10.1007/s12094-024-03842-5.
- 📄 NCCN Guidelines®. Soft Tissue Sarcoma. Versión vigente (2026).
Radioterapia perioperatoria
- 📄 Hayes AJ, et al. UK guidelines for the management of soft tissue sarcomas. Br J Cancer. 2024.
Estadificación
- 📄 Amin MB, Edge SB, Greene FL, et al. AJCC Cancer Staging Manual. 8th ed. Springer; 2017.