Última revisión:
Tumor solitario fibroso
Tumor solitario fibroso (SFT) — definido por fusión NAB2::STAT6. El tumor solitario fibroso es una neoplasia mesenquimal fibroblástica, de comportamiento intermedio (potencial metastásico variable), caracterizada por la fusión NAB2::STAT6 y la positividad nuclear de STAT6 en inmunohistoquímica. Puede aparecer en prácticamente cualquier localización (pleura, retroperitoneo, pelvis, extremidades, cabeza y cuello y, también, meninges).
El término histórico “hemangiopericitoma” se considera obsoleto en la práctica de partes blandas, y hoy se entiende como parte del espectro SFT. El riesgo clínico no se define por el “nombre”, sino por factores de agresividad (tamaño, mitosis, necrosis, edad) y por el resultado de la resección (márgenes).
Puntos críticos (2026): diagnóstico integrado (morfología + IHQ + genética cuando haga falta), estratificación de riesgo, y seguimiento prolongado por recidivas tardías. En enfermedad avanzada, los antiangiogénicos (p. ej. inhibidores tirosina-quinasa) suelen tener más papel que la QT clásica, aunque la estrategia depende de carga tumoral, velocidad de progresión y síntomas.
En 1 minuto
- Qué es: Tumor fibroblástico definido por NAB2::STAT6. IHQ: STAT6 nuclear+ (muy orientativo) y con frecuencia CD34+. Comportamiento intermedio: riesgo de metástasis variable.
- Dónde aparece: Casi en cualquier sitio: pleura, retroperitoneo/pelvis, extremidades, cabeza/cuello y meninges.
- Clínica: Masa de crecimiento lento. Síntomas por efecto masa. Raro pero típico: hipoglucemia (Doege–Potter) por IGF-2.
- Diagnóstico clave: Patrón “patternless” + vasos tipo “staghorn” + colágeno. IHQ: STAT6 nuclear+. Confirmación molecular si duda.
- Tratamiento: Localizado: cirugía R0. RT si alto riesgo o márgenes comprometidos. Avanzado: prioridad frecuente a antiangiogénicos.
- Pitfall: “Hemangiopericitoma” = término legacy. En 2026 manda riesgo (tamaño/mitosis/necrosis/edad) + márgenes + seguimiento largo.
Conceptos
1.1 Definición y nosología (2026)
El SFT es un tumor fibroblástico definido por NAB2::STAT6. El término “hemangiopericitoma” se considera histórico y hoy se engloba en el espectro SFT, especialmente relevante en localizaciones meníngeas por su tendencia a recidiva tardía.
1.2 Epidemiología y localizaciones
- Edad: más frecuente en adultos (amplio rango).
- Topografías: pleura y extrapleural (retroperitoneo/pelvis/extremidades), cabeza y cuello; también meninges.
- Presentación: masa lenta; síntomas por compresión.
1.3 Estratificación de riesgo (práctica)
- Variables usadas habitualmente: tamaño, mitosis, necrosis y edad.
- Alto riesgo → más discusión de RT adyuvante + vigilancia más estrecha.
- Doege–Potter (IGF-2) → pista clínica de SFT grande/activo.
Clínica
El SFT suele ser silente durante años. Los síntomas dependen de la localización y del tamaño. En tumores grandes, puede aparecer hipoglucemia paraneoplásica (síndrome de Doege–Potter) por producción de IGF-2, y de forma más inespecífica dolor, plenitud o síntomas compresivos.
| Localización | Presentación típica | Comentario |
|---|---|---|
| Pleura / intratorácico | Tos, disnea, dolor torácico o hallazgo incidental | Puede alcanzar gran tamaño antes de dar síntomas. |
| Retroperitoneo / pelvis | Plenitud, dolor, compresión urinaria/intestinal | Diagnóstico tardío por espacio de crecimiento. |
| Extremidades | Masa palpable lenta (a veces dolor por compresión) | Profundo suele superar 5 cm al diagnóstico. |
| Cabeza y cuello | Masa, síntomas locales (disfagia, disfonía, obstrucción) | Amplio diferencial; la IHQ es clave. |
| Meninges | Cefalea, crisis, déficit focal | Puede simular meningioma; STAT6 ayuda mucho. |
| Paraneoplásico | Hipoglucemia, a veces hipertrofia osteoartropática | Muy orientativo en tumores grandes/activos. |
* Si hay hipoglucemia sin causa clara, piensa en Doege–Potter y solicita estudio dirigido (glucosa/insulina/C-péptido/IGF-2 según contexto clínico).
Imagen
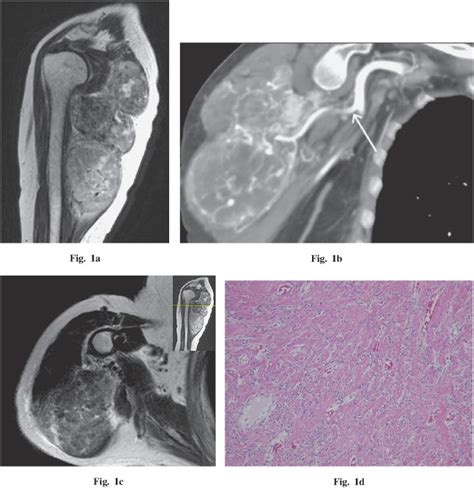
3.1 TC
- Masa de partes blandas, a veces bien delimitada, con realce variable.
- Puede haber necrosis/hemorragia en tumores grandes.
- Útil para estadificación toracoabdominal y plan quirúrgico.
3.2 RM
- Señal variable; áreas hipointensas por colágeno pueden verse.
- Realce tras contraste frecuentemente heterogéneo.
- Clave para relación con planos fasciales, vasos y nervios.
3.3 PET-TC
- Avidez 18F-FDG variable.
- Puede ayudar en sospecha de progresión/recidiva o enfermedad diseminada.
Diagnóstico y diferencial
- Histología típica: patrón “patternless”, alternancia hipo/hipercelular, colágeno, vasos ramificados tipo “staghorn”.
- IHQ: STAT6 nuclear+ (muy orientativo), a menudo CD34+, BCL2+, CD99+. Variable: EMA focal ocasional. Suele ser S-100–, desmina–, citoqueratinas–.
- Molecular: NAB2::STAT6 (RNA-NGS/RT-PCR) si caso equívoco o perfil atípico.
- Agresividad (alertas): necrosis, mitosis altas, pleomorfismo marcado, hipercelularidad difusa, márgenes comprometidos.
- Meningioma (meníngeo): EMA/PR+, STAT6– (útil cuando simula meningioma).
- Sarcoma sinovial: SS18::SSX, citoqueratinas/EMA más consistentes, CD34 habitualmente –.
- DFSP: CD34+, pero patrón/clinica distinta; confirmar por genética (COL1A1-PDGFB) si dudas.
- Schwannoma: S-100/SOX10+.
- Glomangiopericitoma sinonasal: entidad distinta (no es SFT); perfil e historia clínica cambian.
Tratamiento
5.1 Cirugía
Estándar en localizado: resección R0 (márgenes negativos). En meníngeo, resección de inserción dural/hueso según caso. Tumores hipervasculares: considerar planificación hemostática y, en localizaciones seleccionadas, técnicas preoperatorias según equipo.
5.2 Radioterapia
- Adyuvante: alto riesgo (tamaño/mitosis/necrosis/edad) o márgenes R1/R2 si no reintervenible.
- Definitiva: irresecable/recidiva no operable (según localización).
- La decisión es individual: localización, morbilidad, riesgo y preferencia del comité.
5.3 Sistémico (avanzado/metastásico)
- Antiangiogénicos (frecuentes en práctica): pazopanib y otros TKI según línea/contexto clínico.
- QT clásica: antraciclinas ± ifosfamida con respuestas variables (no suele ser el tumor más quimiosensible).
- Opciones en casos seleccionados: combinaciones anti-VEGF/temozolomida o ensayos clínicos.
| Opción | Indicación | Comentario |
|---|---|---|
| Cirugía R0 | Localizado resecable | Base del tratamiento. Márgenes importan. |
| RT adyuvante | Alto riesgo / R1-R2 no rescatable | Decisión por comité según localización y riesgo. |
| Antiangiogénicos (TKI) | Avanzado/metastásico progresivo | Frecuentes en SFT por perfil biológico/vascular. |
| QT (antraciclinas) | Progresión rápida / alta carga / alternativa | Respuesta variable; individualizar. |
Pronóstico y seguimiento
- Riesgo: variable. Se estima con factores como tamaño, mitosis, necrosis y edad.
- Recidiva: puede ser tardía. Esto justifica vigilancia prolongada.
- Metástasis: típicamente pulmón; también hueso e hígado (según series y subgrupos).
- Seguimiento orientativo (ajustar a riesgo/localización):
- 0–3 años: cada 4–6 meses (clínica + imagen del lecho + TC tórax si riesgo).
- 3–10 años: cada 6–12 meses.
- >10 años: anual o cada 1–2 años en alto riesgo (muchos equipos mantienen control largo).
Algoritmo de manejo
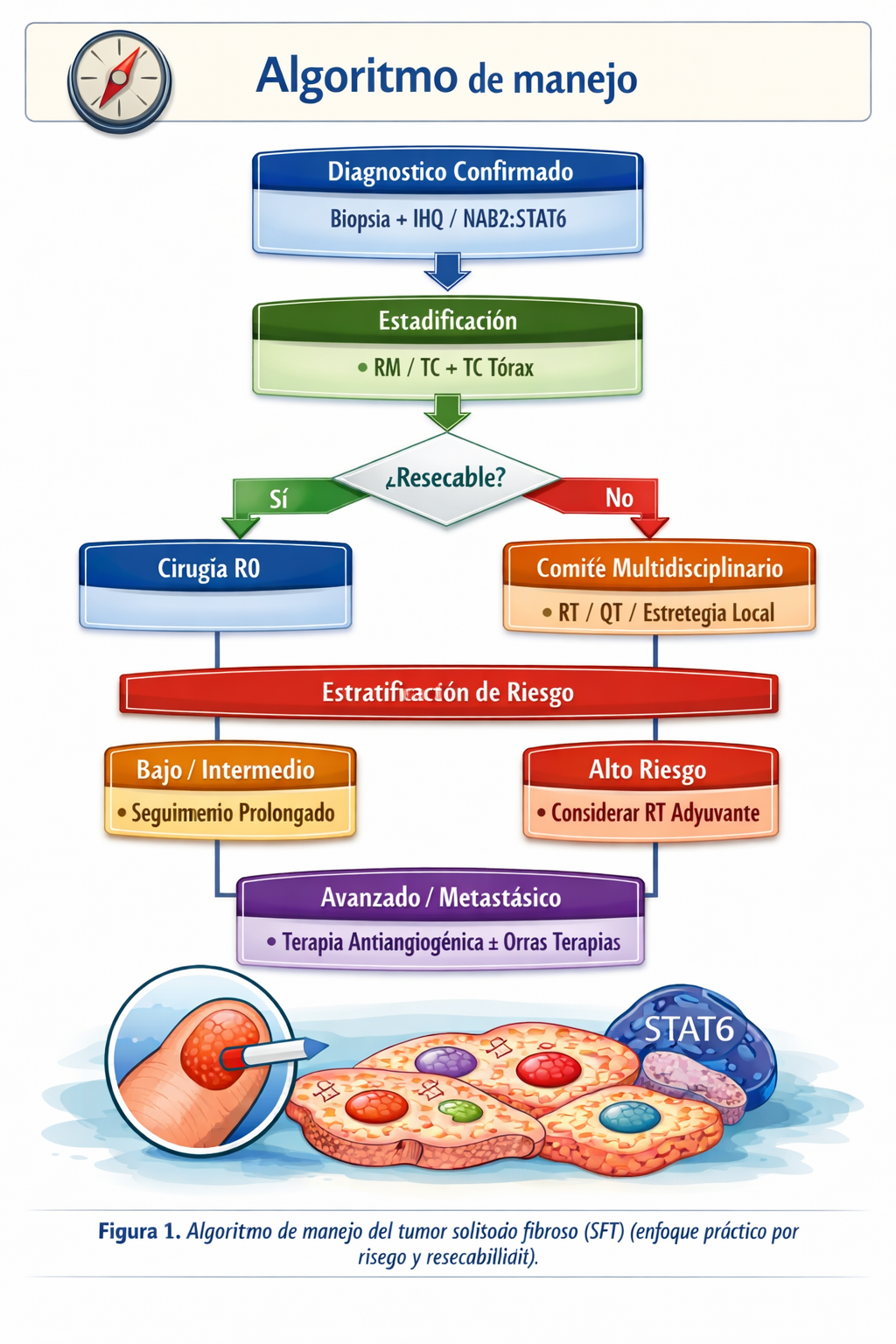
📌 Resumen para la práctica clínica
Indicaciones
- ✓ Resección quirúrgica completa (R0) en enfermedad localizada resecable.
- ✓ Radioterapia adyuvante si alto riesgo o márgenes comprometidos no rescatables.
- ✓ Tratamiento sistémico en enfermedad avanzada/metastásica progresiva o sintomática.
Técnica
- 🔧 Cirugía: resección oncológica con márgenes negativos, preservando función cuando sea posible.
- 🔧 RT: planificación según localización (IMRT/otras técnicas), individualizada en comité.
- 🔧 Sistémico: priorizar estrategia según ritmo de progresión, carga tumoral y perfil del paciente.
Riesgos
- ⚠️ Recidiva local (posible tardía), especialmente si márgenes no son R0.
- ⚠️ Metástasis a distancia en subgrupos de mayor riesgo.
- ⚠️ Complicaciones por efecto masa según localización (compresión neurovascular/visceral).
Resultados
- ✅ El pronóstico es heterogéneo y depende del riesgo histológico-clínico y de los márgenes.
- ✅ Control local alto con R0; el seguimiento prolongado es clave por recaídas tardías.
Bibliografía
Soft Tissue and Bone Tumours. 5th ed. IARC; 2020.
WHO Classification of Tumors of the CNS. 5th ed; 2021.
Recurrent NAB2–STAT6 fusions in solitary fibrous tumor. Nat Genet. 2013.
Solitary fibrous tumor: diagnóstico y entidades del espectro. Mod Pathol. 2014.
Modelos de estratificación de riesgo en SFT. Am J Surg Pathol. 2019.
Guías de sarcomas de partes blandas (últimas actualizaciones).