Tumor de Células Gigantes: Neoplasia Benigna Localmente Agresiva de la Epífisis
El tumor de células gigantes óseo (TCG) es una neoplasia benigna pero localmente agresiva que representa aproximadamente el 5% de todos los tumores óseos primarios y el 20% de los tumores óseos benignos. Afecta típicamente a adultos jóvenes (20-50 años), con una ligera predilección por el sexo femenino, y se localiza característicamente en la región epifisaria de huesos largos, especialmente alrededor de la rodilla (fémur distal, tibia proximal). Aunque es benigno, presenta un comportamiento biológico variable: puede ser localmente destructivo, recidivar tras el tratamiento (tasas del 15-50% según la técnica) y, en raras ocasiones, producir metástasis pulmonares (1-3%) o transformarse malignamente (especialmente tras radioterapia). La Actualización 2026 enfatiza la importancia de la clasificación radiológica de Campanacci (grados I-III) para guiar el tratamiento, el uso de coadyuvantes locales (fenol, criocirugía, fresado de alta velocidad) para reducir las recidivas tras curetaje, y el papel de los fármacos anti-RANKL (denosumab) en el tratamiento neoadyuvante de tumores avanzados o irresecables.
En 1 minuto
- Qué es: El tumor de células gigantes (TCG) es una neoplasia ósea benigna pero localmente agresiva, caracterizada por la presencia de células gigantes multinucleadas tipo osteoclasto en un estroma de células mononucleares. Afecta a adultos jóvenes (20-50 años) y se localiza en la epífisis de huesos largos (fémur distal, tibia proximal, radio distal).
- Clínica típica: Dolor local de larga evolución, tumefacción. Puede debutar con fractura patológica (11-37% de los casos).
- Imagen clave: Lesión lítica excéntrica, metafisoepifisaria, sin matriz calcificada, que adelgaza y expande la cortical. Límites bien definidos pero sin esclerosis periférica. RM: extensión a partes blandas y relación con la articulación.
- Clasificación de Campanacci (grados I-III): Basada en aspectos radiológicos (latente, activo, agresivo). Útil para pronóstico, aunque no predice la recidiva individual.
- Tratamiento: El pilar es la cirugía. El curetaje intralesional amplio + coadyuvantes (fresado de alta velocidad, fenol, criocirugía) + relleno con cemento (PMMA) o injerto óseo es el tratamiento más frecuente, con tasas de recidiva del 10-20%. La resección en bloque está indicada en tumores muy agresivos (grado III), localizaciones reconstructibles (peroné, radio distal) o recidivas. El denosumab (anticuerpo anti-RANKL) se usa como neoadyuvante en tumores avanzados o irresecables.
Conceptos
1.1 Definición
El tumor de células gigantes óseo (TCG) es una neoplasia de bajo grado, caracterizada por la proliferación de células mononucleares del estroma entre las que se distribuyen uniformemente células gigantes multinucleadas de tipo osteoclástico. Aunque histológicamente benigno, tiene capacidad de destrucción ósea local, recidiva tras el tratamiento y, excepcionalmente, puede metastatizar a pulmón (1-3%) o sufrir transformación sarcomatosa (rara).
1.2 Epidemiología
- Frecuencia: Supone aproximadamente el 5% de todos los tumores óseos primarios y el 20% de los tumores óseos benignos (19% en la serie de Schajowicz).
- Edad: Típicamente entre 20 y 50 años (75% de los casos). Raro antes del cierre del cartílago de crecimiento (pico alrededor de los 30 años). En menores de 17 años, más frecuente en mujeres.
- Sexo: Ligera predominancia femenina (especialmente en localizaciones vertebrales).
- Localización:
- Huesos largos (75-90%): Típicamente epífisis (o metáfisis después del cierre fisiario). Más frecuente: fémur distal, tibia proximal, radio distal (alrededor de la rodilla en conjunto).
- Huesos planos y columna: Especialmente sacro y vértebras (en mujeres jóvenes).
- Forma multicéntrica: <1% de los casos. Más agresiva, con predilección por manos y pies, y en pacientes más jóvenes.
1.3 Puntos prácticos (Actualización 2026)
- El diagnóstico diferencial con el tumor pardo del hiperparatiroidismo es obligado: Ante una lesión lítica de células gigantes, debe descartarse hiperparatiroidismo mediante determinación de calcio, fósforo y PTH. El tumor pardo suele asociarse a otras lesiones esqueléticas (osteopenia, resorción subperióstica).
- Clasificación de Campanacci: Útil para describir la agresividad radiológica y orientar el tratamiento, aunque no es predictiva de la recidiva individual. Los grados I (latente) y II (activo) pueden tratarse con curetaje + coadyuvantes; el grado III (agresivo) a menudo requiere resección en bloque.
- Papel del denosumab: El denosumab, un anticuerpo monoclonal anti-RANKL, inhibe la formación y actividad de los osteoclastos, reduciendo la destrucción ósea. Se utiliza como tratamiento neoadyuvante en tumores avanzados, irresecables o en localizaciones complejas (sacro, columna) para facilitar la cirugía. Sin embargo, su uso no está exento de controversia (riesgo de recidiva al suspenderlo, posible aumento de agresividad en recidivas).
- Metástasis pulmonares: Ocurren en el 1-3% de los casos, a menudo tras múltiples recidivas locales. Suelen ser de crecimiento lento. El tratamiento de elección es la resección quirúrgica amplia (metastasectomía). La quimioterapia no ha demostrado ser efectiva.
- Transformación maligna: Rara (menos del 1%). Puede ser espontánea (4-40 años de evolución) o secundaria a radioterapia. La radioterapia con ortovoltaje se asociaba a un alto riesgo; con megavoltaje el riesgo es menor pero no nulo. Por ello, la radioterapia se reserva para casos excepcionales irresecables.
Clínica
| Síntoma/Signo | Frecuencia | Comentario clínico |
|---|---|---|
| Dolor local | >90% | El síntoma más frecuente. Progresivo, de larga evolución. |
| Tumefacción / Masa palpable | Variable | Cuando el tumor expande la cortical o invade partes blandas. |
| Fractura patológica | 11-37% | Forma de presentación en un porcentaje significativo de casos. |
| Síntomas neurológicos | Variable | En localizaciones vertebrales o sacras (compresión radicular). |
| Calcio sérico | Normal | A diferencia del tumor pardo del hiperparatiroidismo. |
Estudios de imagen
3.1 Radiografía simple
- Localización: Lesión lítica, excéntrica, metafisoepifisaria (afecta la epífisis y se extiende a metáfisis).
- Límites: Bien definidos, pero sin esclerosis periférica (a diferencia del fibroma no osificante). Zona de transición estrecha.
- Cortical: Adelgazamiento y expansión, con posible rotura y extensión a partes blandas.
- Matriz: No hay mineralización (ausencia de calcificaciones).
- Espacio articular: Puede estar respetado o invadido en casos avanzados.
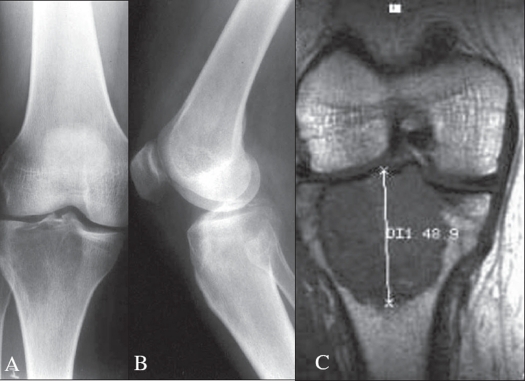
3.2 Resonancia magnética (RM) - Técnica de elección
- Extensión intraósea: Determina la afectación real de la médula.
- Extensión a partes blandas: Evalúa la masa extraósea.
- Relación con la articulación: Evalúa la integridad del cartílago articular y la posible invasión subcondral.
- Señal: Típicamente hipointenso en T1, hiperintenso heterogéneo en T2. Las áreas de hemorragia o quísticas pueden dar señal variable.
3.3 Tomografía computarizada (TC)
- Útil para evaluar el adelgazamiento cortical y la integridad de la cortical.
- Ayuda a planificar la cirugía (especialmente en localizaciones complejas como pelvis o sacro).
3.4 Gammagrafía ósea
- Útil para detectar la forma multicéntrica (<1% de los casos).
Histopatología
4.1 Macroscopía
- Tejido de coloración variable: pardusco, marrón claro, amarillento (por hemosiderina y lípidos).
- Áreas de hemorragia (rojo oscuro) y necrosis (aspecto quístico).
- Consistencia blanda y friable.
4.2 Microscopía
- Células gigantes multinucleadas: Grandes, con numerosos núcleos (20-30), similares a osteoclastos. Distribuidas uniformemente por todo el tumor.
- Células del estroma (mononucleares): Fusiformes u ovaladas, con núcleos redondos u ovalados de tamaño uniforme, cromatina granular y nucléolos prominentes. Son las células neoplásicas verdaderas.
- Mitosis: Pueden verse en las células del estroma (5-10 mitosis por 10 campos de gran aumento). Las células gigantes nunca presentan mitosis.
- No es posible predecir el comportamiento biológico (recidiva, metástasis) basándose solo en la histología.
4.3 Gradación histológica (obsoleta)
Antiguamente se clasificaban en grados I, II y III según el pleomorfismo del estroma, pero este sistema no se correlaciona con el pronóstico y ha sido abandonado.
Clasificación radiológica de Campanacci
Sistema basado en aspectos clínicos y radiológicos, útil para describir la agresividad del tumor y orientar el tratamiento, aunque no es completamente predictivo de la recidiva.
| Grado | Características radiológicas | Comportamiento | Tratamiento típico |
|---|---|---|---|
| I (Latente) | Márgenes bien definidos, cortical intacta, sin extensión a partes blandas. Límite esclerótico. | Indolente, poco sintomático. | Curetaje ± coadyuvantes. |
| II (Activo) | Márgenes relativamente bien definidos, cortical adelgazada y expandida, sin rotura franca. No masa de partes blandas. | Sintomático, crecimiento progresivo. | Curetaje amplio + coadyuvantes + relleno (PMMA/injerto). |
| III (Agresivo) | Márgenes mal definidos, rotura cortical con extensión a partes blandas. Puede invadir la articulación. | Crecimiento rápido, agresivo. | Resección en bloque (amplia) preferible. Curetaje + coadyuvantes en casos seleccionados. |
* No se ha observado correlación entre el grado y la incidencia de recidiva local o metástasis.
Diagnóstico y diagnóstico diferencial
Radiológico:
- Tumor pardo del hiperparatiroidismo: Lesión lítica similar, pero suele acompañarse de otras manifestaciones esqueléticas (osteopenia, resorción subperióstica, pérdida de lámina dura). Calcio/PTH alterados.
- Fibroma no osificante: Localización metafisaria, borde escleroso bien definido, generalmente asintomático.
- Ganglión intraóseo: Lesión lítica con borde escleroso, a menudo yuxtaarticular.
- Quiste óseo aneurismático: Más frecuente en metáfisis de jóvenes, con niveles hidroaéreos. Puede asociarse a TCG (TCG quístico).
- Condroblastoma: Epifisario, pero con calcificaciones y en pacientes más jóvenes (10-20 años).
- Condrosarcoma de células claras: Epifisario, pero con calcificaciones.
- Osteoblastoma: Más frecuente en columna posterior.
- Fibrosarcoma / histiocitoma fibroso maligno: Lesiones líticas agresivas, pero en metáfisis y en pacientes mayores.
- Osteosarcoma fibroblástico o telangiectásico: Lesión lítica agresiva, metafisaria, con posible matriz osteoide.
- Mieloma / metástasis líticas: En pacientes mayores, localización variable.
Patológico (lesiones con células gigantes):
Múltiples lesiones tumorales y pseudotumorales contienen células gigantes. La siguiente tabla resume las principales entidades:
| Tumor o pseudotumor | Tipo de células gigantes |
|---|---|
| Tumor de células gigantes del hueso | Pseudo-osteoclastos y CG muy grandes |
| Osteoma osteoide / Osteoblastoma | CG osteoclásticas |
| Quiste óseo aneurismático | CG osteoclásticas y resortivas |
| Granuloma reparador de células gigantes | CG osteoclásticas y resortivas |
| Quiste óseo simple | CG osteoclásticas y resortivas |
| Condroblastoma | CG "condroclastos" |
| Fibroma condromixoide | CG "condroclastos" |
| Condrosarcoma de células claras | CG osteoclásticas |
| Osteosarcoma rico en células gigantes | Tumor y CG osteoclásticas |
| Osteosarcoma telangiectásico | Tumor CG |
| Fibroma no osificante (defecto fibroso cortical) | CG resortivas |
| Histiocitoma fibroso maligno | Tumor CG |
| Tumor pardo del hiperparatiroidismo | CG osteoclásticas |
| Granuloma de células de Langerhans | CG histiocíticas |
| Enfermedad de Paget | CG osteoclásticas |
Tratamiento
7.1 Opciones quirúrgicas
- Curetaje intralesional amplio + coadyuvantes + relleno: Es el tratamiento más frecuente para la mayoría de los TCG (grados I y II, y algunos III seleccionados).
- Técnica:
- Ventana cortical amplia: Se realiza una ventana suficientemente grande para visualizar toda la cavidad.
- Curetaje agresivo: Extracción de todo el tejido tumoral visible.
- Fresado de alta velocidad: Se utiliza una fresa de alta velocidad para extender el margen mecánicamente y eliminar tumor microscópico de las paredes (reduce recidivas al 12-25%).
- Coadyuvantes locales (opcionales, pero recomendados):
- Fenol (5%): Se aplica en la cavidad durante 1-2 minutos, causando necrosis de 1-2 mm. Tóxico, requiere protección de tejidos blandos. Reduce recidivas al 5-17%.
- Criocirugía con nitrógeno líquido: Se vierte nitrógeno líquido en la cavidad (2-3 ciclos de congelación-descongelación). Penetra 1-2 cm. Tasas de recidiva del 2-12%. Complicaciones: fractura (5.9%), necrosis cutánea (2.9%).
- Coagulación con argón: Alternativa más segura, penetración 2-3 mm. Recidivas del 7% en series iniciales.
- Relleno de la cavidad:
- Cemento de polimetilmetacrilato (PMMA): Ventajas: estabilidad inmediata, permite detectar recidivas precozmente (radiolucencia alrededor del cemento), evita morbimortalidad del injerto. Desventajas: posible daño térmico al cartílago (irrigar con suero frío), dificultad en revisiones.
- Injerto óseo (autólogo o aloinjerto): Restaura el stock óseo, pero con mayor morbilidad y recidivas más difíciles de detectar.
- Técnica:
- Resección en bloque (amplia): Indicada en:
- Tumores grado III con destrucción masiva e invasión articular.
- Localizaciones donde la resección es reconstructible con buena función (peroné proximal, radio distal, clavícula, escápula).
- Recidivas después de curetaje + coadyuvantes.
7.2 Tratamiento médico (denosumab)
- Denosumab: Anticuerpo monoclonal anti-RANKL. Inhibe la maduración y actividad de los osteoclastos.
- Indicaciones:
- Tratamiento neoadyuvante en tumores avanzados, irresecables o en localizaciones complejas (sacro, columna) para facilitar la cirugía.
- Tratamiento de tumores irresecables (control a largo plazo).
- Efectos: Induce esclerosis y osificación del tumor, facilitando el curetaje. Sin embargo, su uso no está exento de controversia: al suspenderlo, puede haber recidiva, y se ha descrito que las recidivas post-denosumab pueden ser más agresivas.
7.3 Radioterapia
- Evitar en lo posible por el riesgo de transformación sarcomatosa (especialmente con técnicas antiguas de ortovoltaje).
- Con técnicas modernas de megavoltaje, el riesgo es menor pero no nulo.
- Reservada para casos excepcionales de tumores irresecables (sacro, columna) en los que la cirugía no es posible.
7.4 Tratamiento de las metástasis pulmonares
- La resección quirúrgica amplia (metastasectomía) es el tratamiento de elección.
- La quimioterapia no ha demostrado eficacia.
- El pronóstico es favorable si se logra la resección completa.
| Técnica | Tasa de recidiva | Comentarios |
|---|---|---|
| Curetaje simple | 35-50% | Inaceptable. No recomendado. |
| Curetaje + fresado alta velocidad | 12-25% | Mejoría significativa. |
| Curetaje + fresado + fenol + PMMA | 5-17% | Estándar actual en muchos centros. |
| Criocirugía (nitrógeno líquido) | 2-12% | Eficaz, pero con riesgo de fractura y necrosis cutánea. |
| Coagulación con argón + PMMA | 7% | Alternativa prometedora (seguimiento corto). |
| Resección en bloque | <10%< /td> | Mayor morbilidad quirúrgica. |
Pronóstico
- Recidiva local: Es la principal complicación. Depende del tratamiento:
- Curetaje simple: 35-50%.
- Curetaje + coadyuvantes (fresado, fenol, criocirugía): 5-20%.
- Resección en bloque: <10%.< /li>
- Metástasis pulmonares: Ocurren en el 1-3% de los casos, generalmente después de múltiples recidivas locales. Suelen ser de crecimiento lento y el tratamiento quirúrgico es curativo en la mayoría.
- Transformación maligna: Rara (<1%). Puede ser espontánea o secundaria a radioterapia. El sarcoma secundario tiene mal pronóstico.
- Factores pronósticos de recidiva:
- Grado de Campanacci (mayor recidiva en grado III).
- Localización (mayor recidiva en radio distal, huesos de la mano).
- Calidad de la cirugía inicial (márgenes, uso de coadyuvantes).
- Presencia de fractura patológica.
Algoritmo de manejo
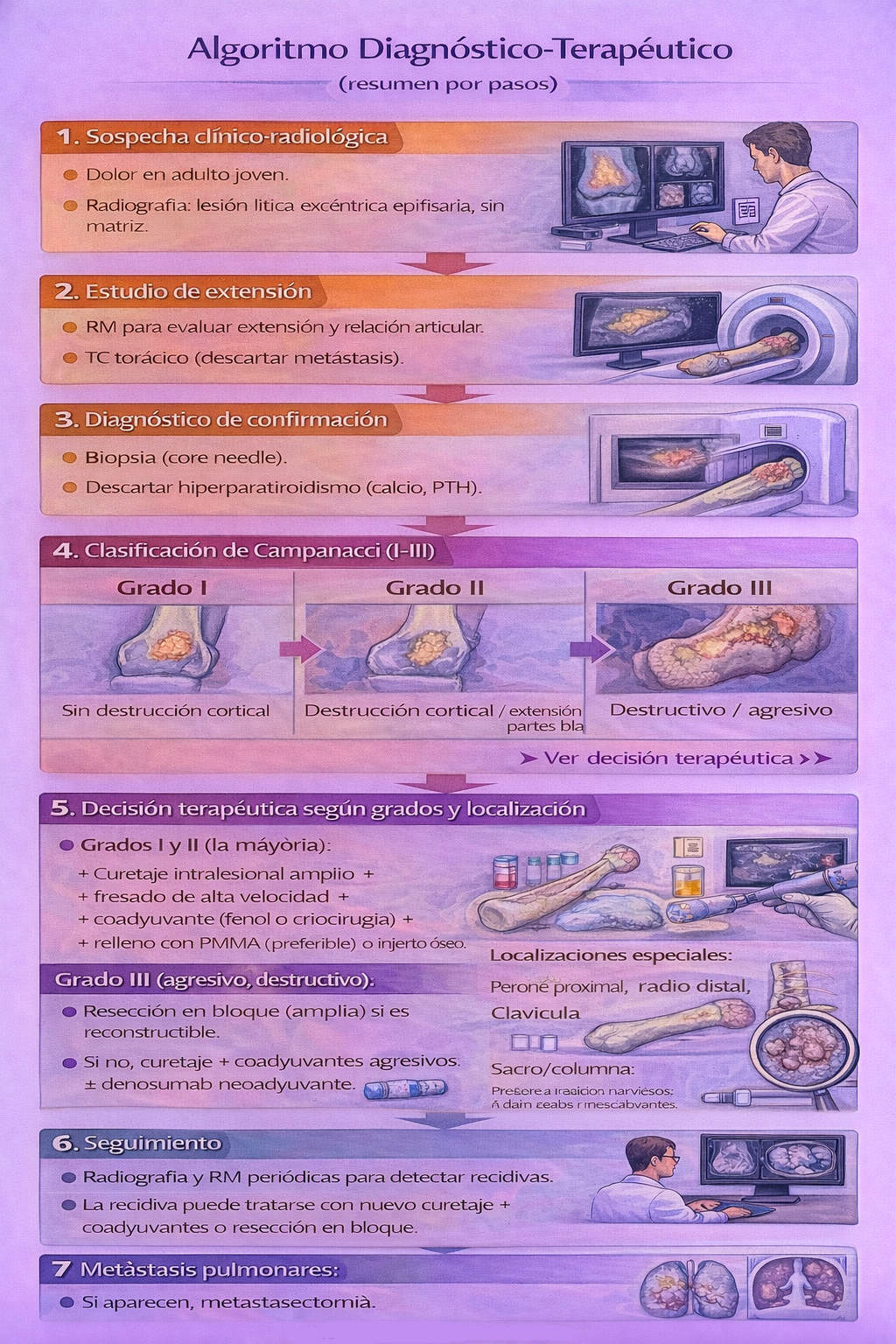
📌 Resumen para la práctica clínica
Indicaciones
- ✓ Curetaje intralesional amplio + fresado de alta velocidad + coadyuvantes (fenol, criocirugía) + relleno con PMMA en tumores de grado I-II.
- ✓ Resección en bloque (amplia) en tumores grado III, localizaciones reconstructibles o recidivas.
- ✓ Denosumab como tratamiento neoadyuvante en tumores avanzados, irresecables o en localizaciones complejas.
Técnica
- 🔧 Curetaje: ventana cortical amplia, curetaje agresivo, fresado de alta velocidad de toda la cavidad.
- 🔧 Coadyuvantes: fenol 5% (1-2 min) o criocirugía (nitrógeno líquido, 2-3 ciclos) con protección de tejidos blandos.
- 🔧 Relleno: PMMA (estabilidad inmediata) o injerto óseo (restaura stock óseo).
Riesgos
- ⚠️ Recidiva local (5-20% con tratamiento óptimo, mayor en grado III y localizaciones difíciles).
- ⚠️ Fractura patológica postoperatoria (especialmente tras criocirugía).
- ⚠️ Infección, no unión, fractura del injerto (en reconstrucciones).
- ⚠️ Metástasis pulmonares (1-3%), generalmente tras múltiples recidivas.
Resultados
- ✅ Tasa de curación definitiva >80% con tratamiento adecuado.
- ✅ Buena función en la mayoría de los casos con cirugía preservadora.
Bibliografía
WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. IARC; 2020.
Giant-cell tumor of bone. J Bone Joint Surg Am. 1987;69(1):106-114.
Cryosurgery in giant cell tumor. Clin Orthop Relat Res. 1999;(359):176-188.
Intralesional excision vs en bloc resection for GCT. J Bone Joint Surg Am. 1993;75(11):1648-1655.
Differential Diagnosis of Tumors and Tumor-Like Lesions of Bones and Joints. Lippincott-Raven; 1998.
Denosumab in giant cell tumour of bone. Lancet Oncol. 2013;14(9):901-908.