Sarcoma Pleomórfico Indiferenciado (UPS) — Actualización 2026: el fin de una denominación histórica y el desafío del diagnóstico de exclusión
El sarcoma pleomórfico indiferenciado (UPS, por sus siglas en inglés) constituye el reemplazo nosológico definitivo del antiguo histiocitoma fibroso maligno (HFM), término que la Organización Mundial de la Salud eliminó en 2013 tras décadas de evidencia acumulada. Lejos de tratarse de un mero cambio de etiqueta, esta reclasificación responde a la demostración de que la inmensa mayoría de las neoplasias previamente diagnosticadas como HFM correspondían en realidad a sarcomas con diferenciación linaje-específica identificable —miogénica, lipoblástica, fibrohistiocítica— o a otros tumores pleomórficos no sarcomatosos. El UPS es hoy un diagnóstico de exclusión: solo puede establecerse tras un estudio inmunohistoquímico y molecular completo que descarte todas las demás entidades. Esta redefinición ha reducido drásticamente su incidencia real, pero no ha modificado su naturaleza: el UPS sigue siendo un sarcoma de alto grado, profundamente agresivo, con marcada tendencia a la recidiva local y a la diseminación hematógena, predominantemente pulmonar. La Actualización 2026 enfatiza la necesidad de centralización en unidades de sarcomas, la obligatoriedad de paneles IHC extensos, la exclusión sistemática de liposarcoma desdiferenciado mediante FISH para MDM2 en localizaciones retroperitoneales, y el reconocimiento de la heterogeneidad pronóstica según órgano de origen, recientemente caracterizada por los grandes registros poblacionales.
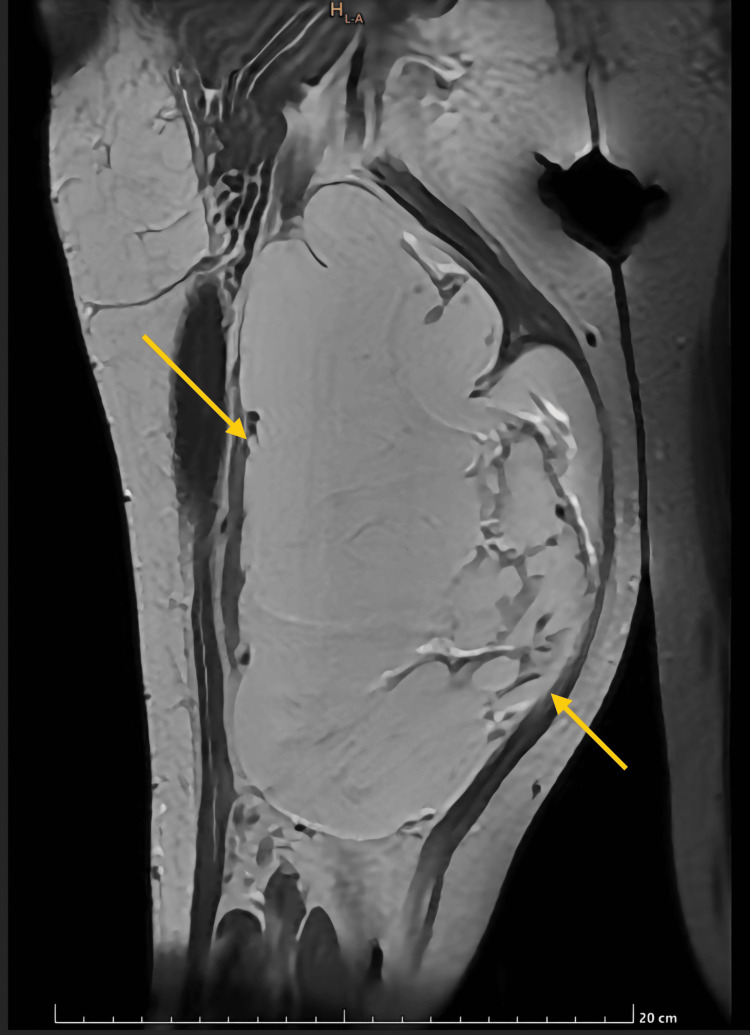
Hallazgo radiológico característico
0) En 1 minuto
- ✅ Qué es: El sarcoma pleomórfico indiferenciado (UPS) es un sarcoma de alto grado, de estirpe mesenquimal, caracterizado por marcado pleomorfismo celular y ausencia completa de diferenciación linaje-específica tras estudio inmunohistoquímico y molecular exhaustivo. La OMS eliminó el término histiocitoma fibroso maligno en 2013; el UPS es hoy un diagnóstico de exclusión .
- ✅ Clínica típica: Masa de crecimiento lento pero progresivo, inicialmente indolora, profunda (fascial o muscular), predominantemente en extremidades (especialmente muslo). El dolor aparece tardíamente por compresión. Menos del 5% se originan en hueso .
- ✅ Imagen clave: Masa bien delimitada en RM pero no encapsulada, tamaño >5 cm al diagnóstico, señal heterogénea en T2, realce intenso heterogéneo, áreas de necrosis/hemorragia. Signos característicos: "fibrous septum sign" (tabiques internos hipointensos), "tail sign" y "pseudocapsule sign". TC torácico obligatorio por alto riesgo metastásico pulmonar .
- ✅ Diferencial clave: El diagnóstico diferencial es el núcleo del proceso. Deben excluirse sistemáticamente: liposarcoma desdiferenciado (MDM2+), leiomiosarcoma (actina+, desmina+), rabdomiosarcoma pleomórfico (miogenina+), melanoma (S100+), carcinoma sarcomatoide (citoqueratinas+). El UPS es negativo para todos los marcadores de linaje .
- ✅ Tratamiento: Cirugía de resección amplia con márgenes negativos (R0) es el estándar. Radioterapia (neo)adyuvante mejora control local en tumores profundos, >5 cm o de alto grado. Quimioterapia con antraciclinas (doxorrubicina) ± ifosfamida en enfermedad metastásica o irresecable; respuesta limitada .
Pitfall: El error más grave y frecuente es no realizar un diagnóstico de exclusión completo. Una neoplasia pleomórfica no puede etiquetarse como UPS sin haber descartado mediante inmunohistoquímica: citoqueratinas (carcinoma), S100 y SOX10 (melanoma, tumor de vaina nerviosa), actina/desmina (sarcoma muscular), MDM2/CDK4 (liposarcoma desdiferenciado) y miogenina/MyoD1 (rabdomiosarcoma). Estudios retrospectivos demuestran que hasta el 70% de los antiguos HFM se reclasifican correctamente al aplicar estos paneles . Asumir el diagnóstico sin esta exclusión conduce a tratamiento subóptimo y errores pronósticos.
1) Conceptos
1.1 Definición y evolución histórica: de HFM a UPS
El término "histiocitoma fibroso maligno" fue acuñado por Stout y Lattes en la década de 1960 y popularizado por Weiss y Enzinger en 1978, quienes describieron una serie de 200 casos de sarcomas pleomórficos que consideraron derivados de histiocitos con capacidad de actuar como fibroblastos facultativos . Durante los siguientes treinta años, el HFM se convirtió en el sarcoma de partes blandas más diagnosticado en adultos, llegando a representar hasta el 20-25% de todos los sarcomas .
Sin embargo, a partir de los años noventa, la acumulación de estudios inmunohistoquímicos y genéticos demostró que la mayoría de estos tumores mostraban en realidad diferenciación miogénica, lipoblástica, fibrohistiocítica o epitelioide. La clasificación de la OMS de 2002 ya introdujo el término "sarcoma pleomórfico indiferenciado" como categoría residual, y en 2013 suprimió definitivamente el término HFM . Actualmente, el UPS se define como un sarcoma de alto grado, compuesto por células pleomórficas fusiformes y redondas, con frecuencia abundantes células gigantes multinucleadas, que no muestra evidencia de diferenciación linaje-específica tras estudio completo .
Esta redefinición no es semántica: tiene consecuencias directas sobre la práctica clínica, el pronóstico y las oportunidades terapéuticas. Un liposarcoma desdiferenciado (MDM2+) puede beneficiarse de terapias dirigidas frente a MDM2; un leiomiosarcoma tiene un comportamiento metastásico diferente; un carcinoma sarcomatoide requiere tratamiento oncológico médico específico. El UPS es, por tanto, el cajón residual tras una criba exhaustiva.
1.2 Epidemiología: una incidencia real muy inferior a la histórica
- 📈 Incidencia real: Extremadamente baja. El UPS supone actualmente menos del 5% de los sarcomas de partes blandas (frente al 20-25% que ocupaba el HFM). Un análisis del registro SEER (2000-2020) identificó solo 1680 casos de sarcomas indiferenciados en dos décadas, con una incidencia estimada de 0.08-1 por 100.000 habitantes/año .
- 📈 Edad y sexo: Pico de incidencia entre 50-70 años (mediana 59,5 años); excepcional en pediatría. Ligero predominio masculino (M:F 1,2:1) sin diferencias pronósticas por sexo .
- 📈 Distribución racial: Más frecuente en caucásicos (64%), seguido de hispanos (15%) y asiáticos (10,5%). La supervivencia es significativamente menor en asiáticos (mediana 1 mes) y negros (mediana 16 meses) frente a caucásicos (mediana 28 meses); diferencias no explicadas por acceso a atención o estadio al diagnóstico .
- 📈 Localización: Extremidades inferiores (~50%), especialmente muslo; extremidades superiores (~20%); tronco y retroperitoneo (15-20%). La localización ósea primaria es rara (1-5%). Los UPS de retroperitoneo deben hacer sospechar siempre liposarcoma desdiferenciado hasta que se demuestre lo contrario .
1.3 Factores de riesgo y etiopatogenia
- 🔹 Radiación: El UPS es el sarcoma radioinducido más frecuente, representando hasta el 60% de los sarcomas postradioterapia. Latencia media: 10-15 años. Riesgo directamente proporcional a la dosis. Especialmente descrito en cáncer de mama, linfoma y cáncer de próstata irradiados .
- 🔹 Antecedentes óseos: Puede surgir sobre lesiones óseas preexistentes: enfermedad de Paget, displasia fibrosa, infarto óseo, osteonecrosis, osteomielitis crónica y quiste óseo aneurismático. En estos casos, el pronóstico es aún más adverso .
- 🔹 Genética molecular: A diferencia de otros sarcomas, el UPS carece de una alteración genética recurrente o diana terapéutica específica. Presenta cariotipos complejos con numerosas alteraciones numéricas y estructurales, pérdida de TP53, RB1, PTEN, y amplificaciones de CCNE1 y ATRX. No hay translocaciones balanceadas ni mutaciones driver identificables .
1.4 Puntos prácticos (Actualización 2026)
- ⚠️ El UPS es un diagnóstico residual: No debe establecerse sin un panel IHC completo y, en casos seleccionados, estudios moleculares (FISH para MDM2, secuenciación). La reclasificación correcta tiene implicaciones pronósticas y terapéuticas directas.
- ⚠️ Supervivencia por localización: El estudio SEER 2024 demuestra que la supervivencia varía drásticamente según órgano de origen: piel/partes blandas: 51 meses; hueso: 32 meses; mama: 30 meses; SNC: 26 meses; tracto gastrointestinal: 10 meses; tracto respiratorio: solo 4 meses. Esta heterogeneidad debe incorporarse a la planificación terapéutica y al consejo pronóstico .
- ⚠️ Papel de la inmunoterapia: Estudios fase II con inhibidores de checkpoint (pembrolizumab, nivolumab) en sarcomas indiferenciados muestran tasas de respuesta bajas (5-10%), inferiores a las observadas en otros sarcomas. No aprobados de forma rutinaria .
- ⚠️ Centralización obligada: Múltiples estudios confirman que la supervivencia es significativamente superior cuando la biopsia, cirugía y tratamiento se realizan en centros de referencia de sarcomas. La cirugía inicial inadecuada (resección intralesional o marginal sin planificación) compromete la posibilidad de cirugía preservadora y aumenta el riesgo de recidiva .
2) Clínica
La presentación clínica del UPS es inespecífica y depende fundamentalmente de la localización, el tamaño y la profundidad del tumor. Dado su crecimiento lento pero sostenido, suele alcanzar dimensiones considerables antes de ser detectado, especialmente en localizaciones profundas como retroperitoneo o muslo.
| Localización | Síntoma principal | Frecuencia | Comentario clínico |
|---|---|---|---|
| Extremidades (profundo) | Masa indolora palpable | >80% | Crecimiento progresivo en meses. Consistencia firme, fija a planos profundos. Piel intacta. Típicamente >5 cm . |
| Extremidades | Dolor local | 25-35% | Por compresión/infiltración nerviosa o muscular. Más frecuente en tumores agresivos o de rápido crecimiento. |
| Extremidades | Claudicación, déficit motor | 10-15% | Afectación de nervio ciático, femoral o plexo braquial. Requiere evaluación funcional preoperatoria . |
| Retroperitoneo | Masa abdominal asintomática | 50-60% | Hallazgo incidental en ecografía/TC. Suele ser >10 cm al diagnóstico. |
| Retroperitoneo | Dolor abdominal/vago | 30-40% | Sensación de plenitud, molestias inespecíficas. El dolor agudo sugiere hemorragia intratumoral. |
| Retroperitoneo | Síntomas compresivos | 20% | Edema en miembros inferiores (compresión venosa), hidronefrosis (compresión ureteral), estreñimiento. |
| Hueso (primario) | Dolor óseo localizado | >90% | Progresivo, nocturno, no mecánico. Puede acompañarse de tumoración palpable . |
| Hueso | Fractura patológica | 15-25% | Forma de presentación en lesiones líticas agresivas, especialmente en metáfisis de fémur o tibia . |
| Cabeza y cuello | Masa cervical, disfagia, disfonía | Variable | Localización infrecuente. El diagnóstico diferencial incluye carcinoma sarcomatoide . |
| Pulmón (primario) | Tos, hemoptisis, disnea | Excepcional | <0,5% de neoplasias pulmonares. Descritos <100 casos. Diagnóstico de exclusión . |
* Los síntomas constitucionales (fiebre, pérdida de peso, anorexia) son infrecuentes y deben hacer sospechar enfermedad metastásica avanzada.
3) Imagen
Los hallazgos de imagen del UPS son inespecíficos y reflejan su naturaleza de sarcoma de alto grado: gran tamaño, márgenes infiltrativos, heterogeneidad y necrosis. La RM es la técnica de elección para la estadificación local; la TC torácica es obligatoria para descartar metástasis.
3.1 Radiografía simple y TC
- 🔹 Radiografía (partes blandas): Masa de tejidos blandos, a menudo sin calcificaciones. En ocasiones, se observan calcificaciones distróficas curvilíneas o puntiformes (15-20% de casos) .
- 🔹 Radiografía (ósea): Lesión lítica agresiva, de bordes mal definidos, permeativa o en "parches", sin matriz osteoide. Puede haber erosión cortical, reacción perióstica (en capas de cebolla, triángulo de Codman) y fractura patológica. No hay osteogénesis tumoral .
- 🔹 TC: Masa de densidad similar al músculo, con áreas hipodensas correspondientes a necrosis, hemorragia o degeneración mixoide. Realce heterogéneo tras contraste. La TC es fundamental para valorar erosión ósea y planificación quirúrgica .
3.2 Resonancia magnética (RM)
- 🔹 Morfología: Masa bien delimitada pero no encapsulada, localizada profundamente al fascia, a menudo en contacto o infiltrando grupos musculares. Tamaño grande (5-20 cm) al diagnóstico .
- 🔹 T1: Señal isointensa o ligeramente hipointensa respecto al músculo. Las áreas de hemorragia subaguda pueden mostrar señal hiperintensa.
- 🔹 T2: Señal marcadamente heterogénea, predominantemente hiperintensa. Las áreas de necrosis y mixoide son muy hiperintensas; las zonas de fibrosis e hipercelularidad son hipointensas. Hasta el 25% corresponden al subtipo mixoide (antes mixofibrosarcoma) .
- 🔹 Hallazgos característicos:
- 🔹 "Fibrous septum sign": Tabiques internos hipointensos en T2 que realzan tras contraste. Refleja bandas fibrosas que dividen el tumor en lóbulos. Altamente sugestivo .
- 🔹 "Tail sign" (signo de la cola): Prolongación lineal del tumor a lo largo de fascias o septos intermusculares. Indica infiltración microscópica más allá del margen aparente .
- 🔹 "Pseudocapsule sign": Anillo periférico hipointenso que no constituye una cápsula verdadera (infiltración microscópica presente).
- 🔹 Edema peritumoral: Frecuente, visible como halo hiperintenso en T2/STIR alrededor del tumor.
- 🔹 T1 con contraste: Realce intenso y heterogéneo de los componentes sólidos. Las áreas de necrosis no realzan. El patrón de realce puede ser periférico o en "panal" .
- 🔹 Difusión (DWI): Restricción en áreas celulares (bajo ADC). Útil para guiar biopsia hacia zonas de mayor grado.
3.3 PET-TC
- 🔹 El UPS es típicamente FDG-ávido (SUVmax elevado, a menudo >5). El SUVmax se correlaciona con grado histológico y proliferación (Ki67). No obstante, su uso rutinario no está recomendado en estadificación inicial. Puede ser útil para detectar recidiva no sospechada o valorar respuesta a quimioterapia neoadyuvante .
3.4 Estadificación metastásica
- ⚠️ TC torácico: OBLIGATORIO en todo diagnóstico de UPS. El pulmón es el sitio predominante de metástasis (70-80%). Hasta el 15-20% de los pacientes tienen metástasis sincrónicas detectables .
- ⚠️ TC abdominopélvico: Indicado en localizaciones de tronco, retroperitoneo o cuando existan síntomas sugestivos. Metástasis hepáticas en 10% de casos avanzados .
4) Diagnóstico y diferencial
Confirmación diagnóstica: El diagnóstico de UPS es exclusivamente histopatológico y requiere biopsia con aguja gruesa (core needle) guiada por imagen, preferiblemente realizada en centro de referencia. La biopsia incisional está indicada solo si la aguja gruesa no es diagnóstica. La aspiración con aguja fina (PAAF) NO es adecuada para diagnóstico inicial.
- 🔬 Histología (HE): Proliferación de células fusiformes y pleomórficas dispuestas en patrón estoriforme (en espiral), fascicular o sólido. Presencia constante de células gigantes multinucleadas de aspecto atípico, figuras mitóticas abundantes (incluyendo atípicas) y áreas de necrosis geográfica. Infiltrado inflamatorio mixto (linfocitos, histiocitos, células xantomatosas) en grado variable .
- 🧪 Inmunohistoquímica (panel obligatorio): El UPS se define por negatividad. Deben incluirse al menos:
- 🔹 Citoqueratinas (AE1/AE3, CAM5.2): negativo → descarta carcinoma sarcomatoide.
- 🔹 S100 y SOX10: negativo → descarta melanoma, tumor de vaina nerviosa.
- 🔹 Actina muscular lisa (SMA), desmina, caldesmón: negativo → descarta leiomiosarcoma, rabdomiosarcoma.
- 🔹 MDM2 y CDK4: negativo → descarta liposarcoma desdiferenciado (especialmente crucial en retroperitoneo).
- 🔹 Miogenina, MyoD1: negativo → descarta rabdomiosarcoma pleomórfico.
- 🔹 CD34, STAT6: negativo → descarta tumor fibroso solitario/hemangiopericitoma.
- 🔹 EMA, MUC4, ETV4: negativos → descartan sarcoma sinovial, sarcoma epitelioide esclerótico.
- 🧬 Genética molecular: No hay alteración específica. El estudio FISH para MDM2 (amplificación) es mandatorio en casos de retroperitoneo y pelvis para excluir liposarcoma desdiferenciado. Pueden observarse mutaciones de TP53, RB1, ATRX, pero no son diagnósticas .
Diagnóstico diferencial (entidades a excluir sistemáticamente):
- 🔹 Liposarcoma desdiferenciado: Principal confundidor, sobre todo en retroperitoneo. IHQ: MDM2+, CDK4+. FISH: amplificación MDM2. Si no se realizan estos marcadores, hasta el 40% de los "UPS" retroperitoneales son en realidad liposarcomas desdiferenciados. Implicación pronóstica y terapéutica (posible respuesta a nutlinas) .
- 🔹 Leiomiosarcoma pleomórfico: SMA+, desmina+, caldesmón+. Genética: pérdida de RB1 y MYOCD. Pronóstico más adverso que UPS puro.
- 🔹 Rabdomiosarcoma pleomórfico: Miogenina+, MyoD1+. Propio de adultos mayores, muy agresivo.
- 🔹 Sarcoma pleomórfico de células fusiformes (NOS): Término genérico cuando no se completa el estudio.
- 🔹 Osteosarcoma pleomórfico de alto grado: Presencia de matriz osteoide; negativo en UPS.
- 🔹 Fibroxantoma atípico: Contrapartida superficial, confinado a dermis, comportamiento indolente. Clave: ausencia de invasión profunda a fascia, presencia de elastosis solar. LN2+ puede verse en ambos .
- 🔹 Melanoma amelánico: S100+, SOX10+, HMB45+, Melan A+.
- 🔹 Carcinoma sarcomatoide: Citoqueratinas+, EMA+. Requiere correlación clínica (antecedente de carcinoma).
5) Tratamiento
Objetivo: Control local duradero con preservación funcional y prevención de metástasis. Dado el alto grado biológico, el tratamiento debe ser agresivo y multidisciplinar, idealmente en centros de referencia de sarcomas.
5.1 Cirugía
- 🔹 Resección amplia (R0): Estándar de tratamiento. Requiere margen de tejido sano de al menos 1-2 cm en plano radial y profundo, o fascia intacta como barrera. La resección intralesional (R2) o marginal (R1) se asocia a tasas de recidiva local >50% .
- 🔹 Cirugía preservadora de extremidad: Actualmente posible en >90% de casos gracias a la combinación con RT neoadyuvante/adyuvante. Requiere planificación conjunta con radioterapeuta y rehabilitador. La amputación queda reservada para casos con infiltración neurovascular masiva no reconstructible o fracaso de cirugías previas .
- 🔹 Biopsia previa: Debe ser realizada por el mismo equipo quirúrgico que hará la resección definitiva. El trayecto de biopsia debe ser excindible en bloque con la pieza quirúrgica. Una biopsia mal planificada puede imposibilitar la cirugía preservadora .
- 🔹 Ganglio centinela: No indicado. La diseminación linfática es excepcional (<5%). La linfadenectomía solo si existe adenopatía clínica o radiológica sospechosa confirmada .
5.2 Radioterapia
- 🔹 Indicaciones: Tumores profundos, >5 cm, alto grado, márgenes positivos o cercanos, localización anatómica que impide resección amplia (cabeza y cuello, columna, base de cráneo, pelvis) .
- 🔹 Neoadyuvante: Preferible en tumores grandes y borderline resecables. Dosis: 50 Gy en fraccionamiento convencional. Reduce el volumen tumoral, facilita márgenes negativos y permite cirugía preservadora. No mejora supervivencia global frente a RT adyuvante, pero asocia menor toxicidad tardía .
- 🔹 Adyuvante: Indicada cuando no se administró RT preoperatoria y hay factores de riesgo. Dosis: 60-66 Gy sobre lecho quirúrgico. Reduce tasa de recidiva local del 30% al 10-15% .
- 🔹 RT exclusiva: Para tumores irresecables. Dosis 70 Gy. Control local subóptimo (30-40% a 5 años).
5.3 Quimioterapia
- 🔹 Neoadyuvante/adyuvante: No hay consenso. No se ha demostrado beneficio en supervivencia global en ensayos aleatorizados. Puede considerarse en tumores de >10 cm, de alto grado o con quimiosensibilidad conocida (no hay predictores). El régimen más utilizado es doxorrubicina 75 mg/m² + ifosfamida 9-10 g/m² .
- 🔹 Enfermedad metastásica: Primera línea: antraciclinas (doxorrubicina sola o combinada). Tasas de respuesta: 15-25%. Segunda línea: si progresión, opciones limitadas: trabectedina (actividad modesta), pazopanib (aprobado en sarcomas de partes blandas, pero eficacia limitada en UPS), gemcitabina + docetaxel (respuestas del 15-20%) .
- 🔹 Inmunoterapia: Pembrolizumab y nivolumab muestran tasas de respuesta <10% en UPS. No aprobados como estándar. Ensayos en curso con combinaciones .
| Opción | Indicación principal | Comentario / Nivel de evidencia |
|---|---|---|
| Cirugía R0 | Todo UPS localizado resecable | Estándar absoluto. La supervivencia a 5 años es del 70-80% si márgenes negativos; cae al 30-40% si márgenes positivos . |
| RT neoadyuvante | Tumores >5 cm, profundos, localización compleja | Control local 85-90% a 5 años. Nivel IA . |
| RT adyuvante | Márgenes positivos/cercanos, recidiva, alto grado | Reduce recidiva local del 30% al 10-15%. Nivel IA . |
| QT (doxorrubicina/ifosfamida) | 1ª línea metastásico; alto riesgo (neoadyuvante) | Respuesta 15-25%. No mejora SG en adyuvancia. Nivel IIB . |
| Pazopanib | 2ª línea metastásico (fracaso a antraciclinas) | Mediana SLE 4-5 meses. Aprobado en sarcomas partes blandas. Nivel IIA . |
| Gemcitabina + docetaxel | 2ª/3ª línea metastásico | Respuesta ~15%. Nivel IIB . |
6) Pronóstico y seguimiento
- 📈 Pronóstico general: El UPS es un sarcoma de alto grado con pronóstico adverso. La supervivencia global a 5 años oscila entre 50-70% en series contemporáneas. Los factores pronósticos independientes son: tamaño (>5 cm), profundidad (profundo vs superficial), localización (extremidad distal vs proximal/axial/retroperitoneal), grado histológico, márgenes quirúrgicos y necrosis a quimioterapia neoadyuvante .
- 📈 Recidiva local: A pesar de tratamiento adecuado, la tasa de recurrencia local a 5 años es del 15-20% (frente al 40-50% antes de la era de la RT). La mayoría ocurren en los primeros 2-3 años, aunque se han descrito recidivas hasta 10 años después .
- 📈 Metástasis a distancia: Es la principal causa de muerte. El 30-50% de los pacientes desarrollarán metástasis, con una mediana de aparición de 12-24 meses. Localización predominante: pulmón (70-80%), seguido de hueso (10-15%), hígado (5-10%) y ganglios linfáticos (<5%) .
- 📈 Seguimiento sugerido (ESMO-NCCN):
- 🔹 Primeros 2-3 años: RM/TC de la región primaria cada 3-4 meses (alto riesgo) o cada 6 meses (bajo riesgo). TC torácico cada 6 meses. Exploración física dirigida.
- 🔹 Hasta 5 años: Cada 6 meses con imagen local y TC torácico.
- 🔹 Hasta 10 años: Anual. El riesgo de metástasis tardías justifica seguimiento prolongado.
- 🔹 Más de 10 años: Individualizar. En tumores de bajo riesgo, alta. En alto riesgo, continuar cada 2 años .
Supervivencia por localización (datos SEER 2024): Es crucial conocer la heterogeneidad pronóstica: piel/partes blandas: 51 meses; hueso: 32 meses; mama: 30 meses; SNC: 26 meses; tracto gastrointestinal: 10 meses; tracto respiratorio: 4 meses. El UPS de origen óseo tiene mejor pronóstico que el osteosarcoma convencional, pero peor que el UPS de partes blandas .
7) Algoritmo
Algoritmo diagnóstico-terapéutico (resumen por pasos)
Biopsia core needle guiada por imagen (RM/TC) en centro de referencia.
Panel obligatorio (citoqueratinas, S100, SOX10, SMA, desmina, MDM2, CDK4, miogenina, MyoD1, CD34, STAT6, EMA).
Imprescindible en localización retroperitoneal/pélvica para excluir liposarcoma desdiferenciado.
RM de región primaria + TC torácico (obligatorio). TC abdominopélvico si procede.
- 🟢 Bajo riesgo: Superficial, <5 cm, resecable R0 previsible.
- 🔴 Alto riesgo: Profundo, >5 cm, retroperitoneal, localización compleja, márgenes dudosos.
- ✅ Bajo riesgo: Cirugía R0. Si márgenes negativos → observación.
- ✅ Alto riesgo resecable: Valorar RT neoadyuvante (50 Gy) → cirugía R0 → completar RT si márgenes positivos.
- ✅ Alto riesgo irresecable: RT definitiva (70 Gy) ± QT neoadyuvante (individualizar).
- 🔹 1ª línea: doxorrubicina ± ifosfamida.
- 🔹 2ª línea: pazopanib, gemcitabina + docetaxel, trabectedina (escasa evidencia).
- 🔹 Oligometástasis pulmonares: metastasectomía si control del primario.
Guarda el archivo como images/algoritmo_sarcoma_pleomorfico_indiferenciado.(svg/png/jpg) para mostrar el diagrama de flujo.
8) UPS primario de hueso (sarcoma pleomórfico indiferenciado óseo)
En la clasificación moderna, el equivalente óseo del antiguo “malignant fibrous histiocytoma of bone (MFH)” se integra como sarcoma pleomórfico indiferenciado de alto grado del hueso (denominación usada en revisiones radiológicas de la clasificación OMS). Es una entidad rara, sin producción de osteoide tumoral, y su abordaje terapéutico suele seguir los esquemas de osteosarcoma de alto grado (quimioterapia multiagente + resección amplia). :contentReference[oaicite:1]{index=1}
8.1 Cuándo hablar de UPS de hueso (criterio práctico)
- ⚠️ Requisito clave: Tumor pleomórfico de alto grado en hueso sin matriz osteoide tumoral y tras excluir entidades “mimicker” (osteosarcoma pleomórfico, fibrosarcoma de hueso, metástasis carcinoma sarcomatoide, melanoma, etc.).
- ⚠️ Terminología: En revisiones ligadas a la OMS se describe como sustitución del término “MFH” por undifferentiated high-grade pleomorphic sarcoma of bone. :contentReference[oaicite:3]{index=3}
8.2 Clínica e imagen (resumen estructurado)
- 🔹 Clínica típica: Dolor óseo progresivo (frecuente nocturno), tumefacción y, en un subgrupo, fractura patológica.
- 🔹 Rx/TC: Lesión lítica agresiva con amplia zona de transición; destrucción cortical y componente de partes blandas.
- 🔹 RM: Reemplazo medular con extensión extraósea; patrón inespecífico de sarcoma de alto grado. En revisiones radiológicas se describe señal T1 iso- a discretamente hiperintensa respecto a músculo y T2 discretamente hiperintensa, con agresividad marcada. :contentReference[oaicite:4]{index=4}
8.3 Diagnóstico y diferencial (óseo)
- 🔹 Objetivo: Confirmar sarcoma de alto grado y demostrar ausencia de osteoide tumoral (si hay osteoide → osteosarcoma pleomórfico).
- 🔹 Diferencial prioritario: osteosarcoma pleomórfico, fibrosarcoma de hueso (diagnóstico de exclusión), sarcoma postradiación, metástasis (carcinoma sarcomatoide), melanoma, linfoma óseo.
- 🔹 Nota práctica: En el marco PDQ (NCI), se considera dentro del bloque “osteosarcoma y UPS de hueso” y se trata de forma similar al osteosarcoma, precisamente por su comportamiento y manejo oncológico.
8.4 Tratamiento (orientación clínica estándar)
- ⚠️ Principio: Manejo tipo osteosarcoma de alto grado: quimioterapia multiagente + resección amplia del tumor primario. :contentReference[oaicite:6]{index=6}
- ⚠️ Cirugía: Resección amplia (en bloque) con reconstrucción según localización.
- ⚠️ QT: Esquemas y estrategia (neo/adyuvante) según protocolos de sarcoma óseo de alto grado (centro de referencia).
- ⚠️ RT: Individualizar; más habitual como consolidación si resección incompleta/no resecable o control local específico (según comité).
Para tu índice: crea la entrada “UPS (hueso)” apuntando a sarcoma_pleomorfico.php#ups-hueso.
Resumen para la práctica clínica
📌 Indicaciones
- ✅ Partes blandas: resección quirúrgica amplia con márgenes negativos (R0) en todo UPS localizado.
- ✅ Partes blandas: radioterapia (neo)adyuvante en tumores de alto riesgo (profundos, >5 cm, márgenes comprometidos o localización anatómica desfavorable).
- ✅ Hueso: manejo tipo osteosarcoma de alto grado (quimioterapia multiagente + resección amplia) en UPS primario óseo.
🔧 Técnica
- 🔧 Partes blandas: resección En bloc con margen de 1-2 cm o fascia intacta; planificación conjunta con radioterapia cuando proceda.
- 🔧 Partes blandas: radioterapia IMRT/protones según localización. Neoadyuvante: 50 Gy; adyuvante: 60-66 Gy.
- 🔧 Hueso: enfoque de sarcoma óseo de alto grado con cirugía oncológica y quimioterapia según protocolos del centro.
⚠️ Riesgos
- ⚠️ Recidiva local (15-20% a 5 años con tratamiento óptimo en partes blandas).
- ⚠️ Metástasis a distancia, especialmente pulmonares (30-50% de los pacientes).
- ⚠️ Complicaciones quirúrgicas y funcionales (especialmente en resecciones complejas o reconstrucciones óseas).
- ⚠️ Toxicidad por radioterapia (fibrosis, edema, segundos tumores) cuando se indica.
- ⚠️ Respuesta limitada a QT en enfermedad avanzada de partes blandas; en hueso, la estrategia QT es protocolizada tipo osteosarcoma.
✅ Resultados
- ✅ Supervivencia global a 5 años (global): 50-70% (muy dependiente de tamaño, márgenes y localización).
- ✅ Control local en partes blandas: 80-85% tras cirugía R0 + RT cuando procede.
- ✅ En UPS óseo, pronóstico y tratamiento se abordan dentro de protocolos de sarcoma óseo de alto grado (referencia a comité especializado).
9) Bibliografía
Referencias clave (clasificación OMS, epidemiología, guías)
- 📄 Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer. 2014;120(12):1763-1774.
- 📄 Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon: IARC Press; 2020.
- 📄 Ali H, Prabhakaran SY, Saleem HMA, Thirumaran R, Desai K. Epidemiology of undifferentiated sarcomas: A rare entity. J Clin Oncol. 2024;42(16_suppl):e23571.
- 📄 von Mehren M, Kane JM, Agulnik M, et al. Soft Tissue Sarcoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024;22(4):249-266.
- 📄 ESMO-EURACAN Clinical Practice Guidelines. Soft tissue and visceral sarcomas. Ann Oncol. 2021;32(4):457-470.
- 📄 Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978;41(6):2250-2266.
Radiología y diagnóstico por imagen
- 📄 Park SW, Kim HJ, Lee JH, Ko YH. Malignant fibrous histiocytoma of the head and neck: CT and MR imaging findings. AJNR Am J Neuroradiol. 2009;30(1):71-76.
- 📄 Karki B, Xu YK, Wu YK, Zhang WW. Primary malignant fibrous histiocytoma of the abdominal cavity: CT findings and pathological correlation. World J Radiol. 2012;4(4):151-158.
- 📄 Razek AA, Huang BY. Soft tissue tumors of the head and neck: imaging-based review of the WHO classification. Radiographics. 2011;31(7):1923-1954.
Tratamiento y pronóstico
- 📄 Kilitçi A, Yilmaz F, Kilicgun H, Uygun N. A Case of Primary Undifferentiated Pleomorphic Sarcoma of the Lung. J Coll Physicians Surg Pak. 2021;31(11):1380-1381.
- 📄 Vodanovich DA, M Choong PF. Soft-tissue sarcomas. Indian J Orthop. 2018;52(1):35-44.
- 📄 Gronchi A, Miah AB, Dei Tos AP, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(11):1348-1365.
- 📄 Yang Y, Xu L, Li Y, Miao J. Clinical characteristics and prognosis of undifferentiated pleomorphic sarcoma: a population-based study using SEER database. Front Oncol. 2023;13:1148254.
Fisiopatología y genética
- 📄 Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017;171(4):950-965.e28.
- 📄 Steele CD, Tarabichi M, Oukrif D, et al. Undifferentiated Sarcomas Develop through Distinct Evolutionary Pathways. Cancer Cell. 2019;35(3):441-456.e8.
UPS de hueso (terminología y manejo)
- 📄 NCI PDQ. Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment (summary). (Consultado como referencia de manejo tipo osteosarcoma).
- 📄 Revisiting the WHO classification system of bone tumours (review radiológica).