Sarcoma de Ewing (óseo y extraóseo) — Actualización 2026: entidad OMS/WHO (2020+) definida por fusión EWSR1/FUS::ETS (p. ej., EWSR1::FLI1), alta quimiosensibilidad y pronóstico dependiente de estadio, localización y respuesta
El sarcoma de Ewing es un sarcoma de células redondas pequeñas de alto grado, típicamente en niños, adolescentes y adultos jóvenes. En la clasificación OMS/WHO actual, los términos “pPNET” y “tumor de Askin” se consideran históricos y se agrupan como sarcoma de Ewing (de hueso o de partes blandas). La mayoría de casos presentan una fusión EWSR1::ETS, especialmente EWSR1::FLI1 (~85–90%) y EWSR1::ERG (~5–10%); raramente existen fusiones con FUS::ETS u otros socios ETS. El diagnóstico actual exige confirmación molecular (idealmente panel de fusiones por NGS/RNA; FISH/RT-PCR según disponibilidad), porque múltiples tumores “Ewing-like” pueden imitarlo (CIC-rearranged, BCOR-altered, EWSR1-non-ETS como NFATC2/PATZ1) y requieren enfoque diferenciado.
El tratamiento estándar de primera línea es multimodal: quimioterapia sistémica tipo VDC/IE (vincristina, doxorrubicina, ciclofosfamida alternando con ifosfamida/etopósido), preferiblemente interval-compressed (cada 2 semanas con G-CSF), seguida de control local (cirugía y/o radioterapia) y completar quimioterapia. La supervivencia en enfermedad localizada suele situarse alrededor del 70–80%, y en enfermedad metastásica cae de forma marcada (mejor si metástasis exclusivamente pulmonares).
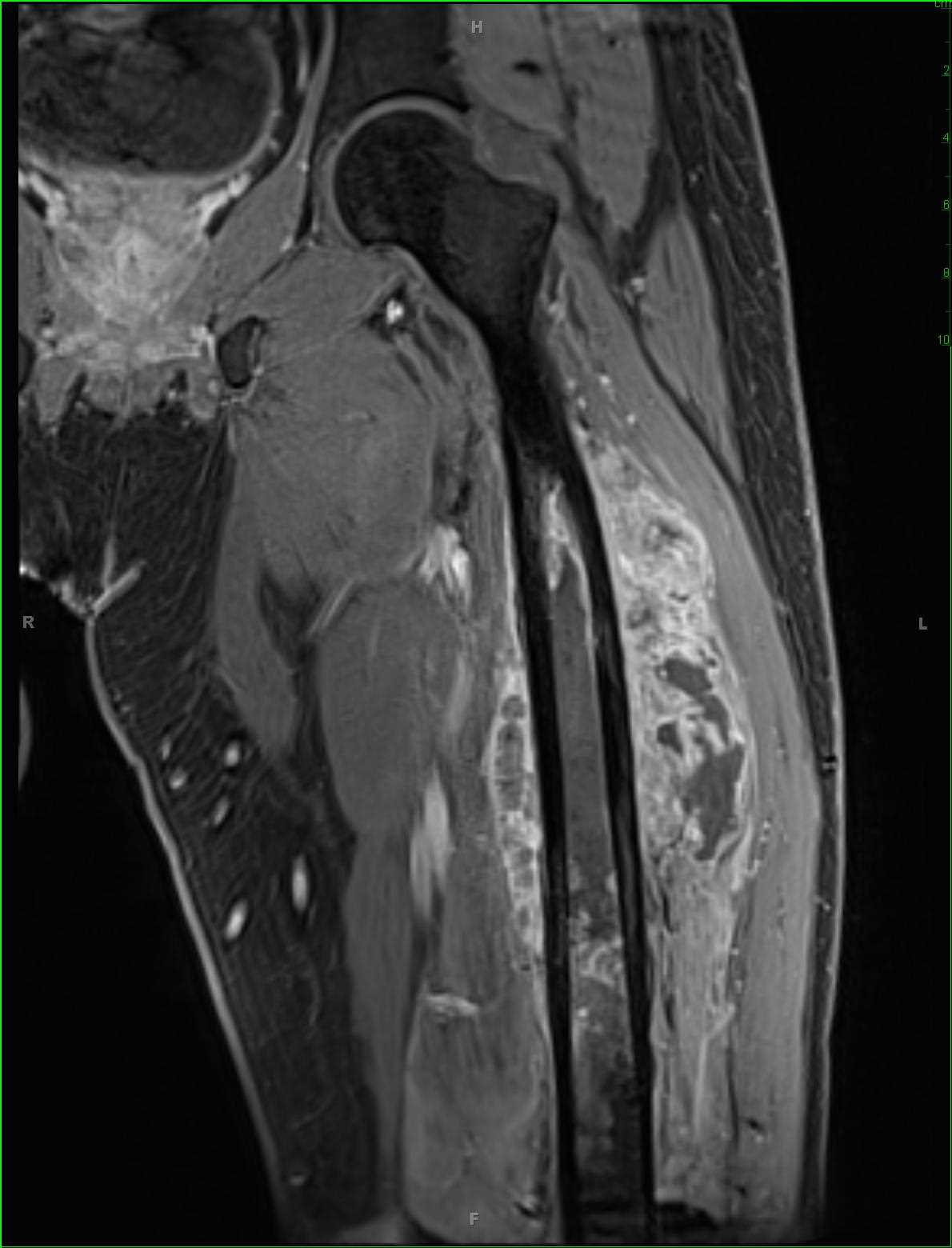
Hallazgo radiológico característico
0) En 1 minuto
- ✅ Qué es: Sarcoma de Ewing (óseo o extraóseo) = sarcoma de células redondas pequeñas de alto grado definido por fusión EWSR1/FUS::ETS (principalmente EWSR1::FLI1). “pPNET/Askin” se consideran términos históricos.
- ✅ Clínica típica: dolor + masa de rápido crecimiento; puede asociar fiebre, ↑VSG/↑LDH, simulando osteomielitis. Pico 10–20 años.
- ✅ Imagen clave: Rx con lesión agresiva permeativa y reacción perióstica laminar; RM para extensión local. TC torácico siempre. PET-TC muy útil para metástasis óseas y respuesta.
- ✅ Diagnóstico: biopsia planificada + IHQ orientativa (CD99 patrón membranoso; NKX2.2 nuclear) pero el paso crítico es confirmación molecular de la fusión (idealmente NGS/RNA; alternativa FISH/RT-PCR).
- ✅ Tratamiento: siempre multimodal: QT VDC/IE (mejor interval-compressed) → control local (cirugía ± RT) → completar QT. En localizado, resultados globales ~70–80%.
Pitfall: No basta con morfología + CD99. CD99 no es específico y existen sarcomas “Ewing-like” (CIC-rearranged, BCOR-altered, EWSR1-non-ETS) que pueden ser CD99+ y requieren confirmación molecular. Si solo usas IHQ básica, el riesgo de diagnóstico erróneo es real y cambia el manejo.
1) Conceptos y clasificación
1.1 Definición (OMS/WHO)
El sarcoma de Ewing es un sarcoma de células redondas pequeñas definido por una fusión entre un gen de la familia FET (habitualmente EWSR1; raramente FUS) y un factor de transcripción de la familia ETS (sobre todo FLI1 o ERG). Puede originarse en hueso o partes blandas. Los términos “pPNET” y “tumor de Askin” se consideran nomenclatura histórica (misma entidad biológica).
1.2 “Sarcomas de células redondas” y diferenciales moleculares (clave 2026)
Además del sarcoma de Ewing, la OMS/WHO reconoce (entre otros) estos grandes grupos que pueden simularlo:
- 🔹 Round cell sarcoma con EWSR1::non-ETS (p. ej., NFATC2, PATZ1): fenotipo “Ewing-like” pero biología distinta.
- 🔹 Sarcoma CIC-rearranged (p. ej., CIC-DUX4): más agresivo, menor quimiosensibilidad.
- 🔹 Sarcoma con alteraciones BCOR (p. ej., BCOR::CCNB3): puede parecer Ewing; IHQ y fusión son determinantes.
Mensaje práctico: ante tumor de células redondas, el objetivo no es “CD99+”, sino definir la fusión (RNA-NGS si es posible).
1.3 Nota histórica: “Familia de tumores de Ewing (EFT)”
En textos clásicos se describía “EFT” (Ewing óseo, extraóseo, pPNET, Askin). Para 2026, puede mencionarse como contexto histórico, pero la entidad diagnóstica recomendada es sarcoma de Ewing.
2) Epidemiología
- 📈 Edad: pico en la 2ª década (10–20 años). Raro <3 años y menos frecuente en mayores.
- 📈 Incidencia: tumor raro (≈1–3 por millón/año). En pediatría/adolescencia es el 2º sarcoma óseo maligno tras osteosarcoma.
- 📈 Sexo: ligero predominio masculino.
- 📈 Ascendencia: incidencia claramente mayor en población de ascendencia europea que en ascendencia africana o asiática.
- 📈 Origen: ≈80–85% óseo; ≈15–20% extraóseo (partes blandas).
Las tablas clásicas por “subtipo pPNET/Askin” tienen valor histórico, pero hoy se recomienda informar como “sarcoma de Ewing” y añadir localización (óseo/extraóseo) y fusión.
3) Localización
Puede aparecer en cualquier sitio. En hueso predomina en pelvis y huesos largos; en extraóseo puede aparecer en pared torácica, paraespinal, retroperitoneo, etc.
* La afectación axial (pelvis/columna) se asocia a peor control local y pronóstico.
4) Clínica
| Síntoma/Signo | Frecuencia | Comentario clínico |
|---|---|---|
| Dolor localizado | >90% | Suele ser inicial; puede empezar intermitente y progresar. |
| Masa/tumefacción | 70–80% | Rápido crecimiento, a veces muy voluminosa. |
| Síntomas sistémicos | 20–30% | Fiebre, ↑VSG/↑LDH, leucocitosis. Puede simular osteomielitis. |
| Fractura patológica | 5–10% | Menos frecuente que en otros tumores óseos. |
| Síntomas neurológicos | Raro | Compresión medular si vertebral/paraespinal. |
Al diagnóstico, ~20–30% presentan metástasis visibles (pulmón, hueso, médula ósea); la mayoría tienen micrometástasis subclínicas.
5) Estudios de imagen
5.1 Radiografía
- 🔹 Lesión agresiva lítica mal definida, patrón permeativo.
- 🔹 Reacción perióstica laminar (“capas de cebolla”) ± espículas.
- 🔹 Masa de partes blandas frecuente.
5.2 RM (imprescindible)
- 🔹 Elección para estadificación local: extensión intramedular, partes blandas, paquete neurovascular, articulación.
- 🔹 Recomendable RM de todo el hueso afecto (buscar “skip lesions” en el mismo hueso).
5.3 Estadificación a distancia
- 🔹 TC torácico: obligatorio (metástasis pulmonares).
- 🔹 PET-TC (FDG): muy útil para metástasis óseas/partes blandas y respuesta.
- 🔹 Gammagrafía ósea: alternativa si no hay PET-TC, pero cada vez menos usada como primera opción.
- 🔹 Biopsia de médula ósea: puede ser selectiva (p. ej., si hay sospecha/metástasis en PET-TC o alto riesgo); en algunos consensos puede omitirse si PET-TC y resto de estudio son negativos.
6) Histopatología
6.1 Macroscopía
- 🔹 Masa blanda “carnosa”, gris-blanquecina, con necrosis/hemorragia.
- 🔹 En hueso suele acompañarse de gran componente de partes blandas.
6.2 Microscopía (HE)
- 🔹 Proliferación difusa de células pequeñas y uniformes.
- 🔹 Citoplasma escaso; frecuentemente con glucógeno (PAS+, diastasa-PAS sensible).
- 🔹 Tabiques fibrovasculares finos; necrosis frecuente.
7) Inmunohistoquímica (orientativa, no definitiva)
CD99 suele ser fuerte y membranoso, pero no es específico. Marcadores útiles adicionales: NKX2.2 (nuclear, sensible pero no perfecto), FLI1/ERG (nuclear, con solapamientos).
Diagnóstico definitivo: fusión EWSR1/FUS::ETS por método molecular.
| Entidad | IHC típica (atajos útiles) | Clave confirmatoria |
|---|---|---|
| Sarcoma de Ewing | CD99 membranoso difuso; NKX2.2 nuclear frecuente; queratinas/EMA generalmente negativas o focales | EWSR1/FUS::ETS (FLI1/ERG/ETV/FEV) |
| Linfoma | CD45 (LCA)+; marcadores B/T según subtipo | Inmunofenotipo linfoide |
| Rabdomiosarcoma | Desmina+, Myogenin/MyoD1+ | Fusión FOXO1 en alveolar (si aplica) |
| Sarcoma sinovial (pobremente diferenciado) | Queratinas/EMA a veces +; TLE1+ (orienta) | SS18::SSX |
| DSRCT | Desmina “dot”; WT1 (C-term)+; queratinas+ | EWSR1::WT1 |
| CIC-rearranged | CD99 variable; NKX2.2 habitualmente −; ETV4 puede ayudar | CIC rearrangement (p. ej., CIC-DUX4) |
| BCOR-altered (BCOR::CCNB3) | BCOR+, CCNB3+, SATB2+ (a menudo); CD99 variable | BCOR::CCNB3 u otras alteraciones BCOR |
| Neuroblastoma/metástasis | Sinaptofisina/cromogranina+; PHOX2B+; CD99 suele − | Contexto clínico + genética (p. ej., MYCN) |
| Osteosarcoma de células pequeñas | SATB2+ puede orientar; CD99 puede ser +; buscar osteoide | Osteoide tumoral (histología) |
8) Patología molecular
8.1 Fusiones características
- 🔹 EWSR1::FLI1 (~85–90%).
- 🔹 EWSR1::ERG (~5–10%); en algunos casos puede ser difícil con ciertos FISH break-apart por complejidad estructural.
- 🔹 Raras: EWSR1::ETV1, EWSR1::ETV4, EWSR1::FEV, y fusiones con FUS::ETS (p. ej., FUS::ERG/FEV).
Implicación práctica: si FISH es negativo pero la sospecha es alta, prioriza RNA-NGS (panel de fusiones).
8.2 Métodos recomendados (2026)
- 🔹 RNA-NGS (panel de fusiones): muy útil para ETS y no-ETS, y para “Ewing-like”.
- 🔹 FISH: rápido; pero puede no resolver socio y puede fallar en variantes complejas.
- 🔹 RT-PCR: sensible si conoces la fusión; menos flexible ante variantes.
8.3 Alteraciones adicionales (estratificación/riesgo)
- 🔹 Alteraciones secundarias (p. ej., CDKN2A, STAG2, TP53) pueden asociarse a peor evolución en algunas series, pero no sustituyen al estadio/respuesta.
9) Diagnóstico y diagnóstico diferencial
Confirmación diagnóstica (mínimo 2026):
- ✅ Biopsia (core/incisional) planificada con trayecto excindible.
- ✅ Histología + panel IHQ de células redondas.
- ✅ Estudio molecular de fusión (preferible RNA-NGS; alternativa FISH/RT-PCR).
- ✅ Conservar material (bloques y, si se puede, congelado) para pruebas adicionales.
Diferencial prioritario 2026:
- 🔹 Ewing-like: CIC-rearranged, BCOR-altered, EWSR1-non-ETS (NFATC2/PATZ1).
- 🔹 Sinovial pobremente diferenciado (SS18::SSX).
- 🔹 DSRCT (EWSR1::WT1).
- 🔹 Linfoma (CD45+).
- 🔹 Rabdomiosarcoma (myogenin/MyoD1+).
- 🔹 Osteosarcoma de células pequeñas (buscar osteoide; SATB2 orienta).
Regla: si es “tumor de células redondas”, define la fusión.
10) Estadificación
- 📍 Localizada: sin metástasis a distancia.
- 📍 Metastásica: pulmón/pleura, hueso, médula ósea, ganglios, SNC (raro).
Recomendado: RM local (incluyendo todo el hueso), TC torácico y PET-TC (o equivalente). BMAB puede ser selectiva según protocolo/riesgo.
Nota: “skip lesions” dentro del mismo compartimento/hueso se consideran extensión loco-regional en varios protocolos, no metástasis sistémica.
11) Pronóstico y factores pronósticos
- 📊 Localizado: SG 5 años típicamente ~70–80% con tratamiento moderno.
- 📊 Metastásico: peor; mejor si metástasis solo pulmonares que si óseas/médula.
Factores clave: estadio al diagnóstico, volumen/tamaño, localización axial (pelvis/columna), LDH, y sobre todo respuesta a QT (necrosis/viabilidad tumoral en pieza quirúrgica o respuesta metabólica).
La asociación pronóstica de “tipo 1” EWSR1::FLI1 se ha descrito históricamente, pero en 2026 no suele usarse para decisiones terapéuticas fuera de investigación.
12) Tratamiento
Premisa: manejo en centros de referencia, siempre multimodal y con planificación por comité.
12.1 Quimioterapia (1ª línea)
- 🔹 Esquema estándar ampliamente aceptado (2026): VDC/IE alternante, idealmente cada 2 semanas (interval-compressed) con soporte G-CSF.
- 🔹 Secuencia típica: ~6 ciclos de inducción → control local → completar hasta ~14 ciclos totales (puede variar por protocolo).
- 🔹 Objetivos: tratar micrometástasis, reducir primario, y medir respuesta.
12.2 Control local: cirugía y/o radioterapia
- 🔹 Cirugía si resección amplia R0 y función aceptable.
- 🔹 RT si irresecable, márgenes positivos no rescatables o localizaciones complejas (pelvis/columna).
- 🔹 Dosis orientativas: 45–50.4 Gy postoperatoria (según márgenes) y ~54–55.8 Gy definitiva (fracciones 1.8 Gy), ajustando a protocolo/volumen.
- 🔹 Técnicas: IMRT/protones cuando aporten reducción de toxicidad.
12.3 Metástasis pulmonares (WLI) y control de metástasis
- 🔹 WLI tras respuesta a QT en metástasis pulmonares: rangos habituales 15–18 Gy, con esquemas dependientes de edad/protocolo (p. ej., 15 Gy/10 fr si <14 años; 18 Gy/12 fr si ≥14 años) y boost si lesión macroscópica residual.
- 🔹 Metástasis óseas/partes blandas: considerar RT dirigida/ablativa en lesiones seleccionadas según protocolo.
12.4 Recaída / refractario (resumen práctico)
- 🔹 Prioridad: ensayo clínico si disponible.
- 🔹 QT de rescate: topotecán/ciclofosfamida; irinotecán/temozolomida ± vincristina; ifosfamida a dosis altas; gemcitabina/docetaxel (según caso/edad/centro).
- 🔹 Terapias dirigidas con evidencia de actividad modesta en series/ensayos: regorafenib, cabozantinib (selección individual y toxicidad).
| Escenario | Tratamiento (esquema general) | Pronóstico |
|---|---|---|
| Localizado | QT VDC/IE → control local → completar QT | Mejor (≈70–80%) |
| Metastásico solo pulmón | QT → control local + WLI ± resección/boost | Intermedio |
| Metastásico óseo/médula | QT intensiva + control local + RT metástasis selectiva | Peor |
| Recaída | Ensayo clínico o QT rescate ± cirugía/RT | Variable |
13) Algoritmo de manejo
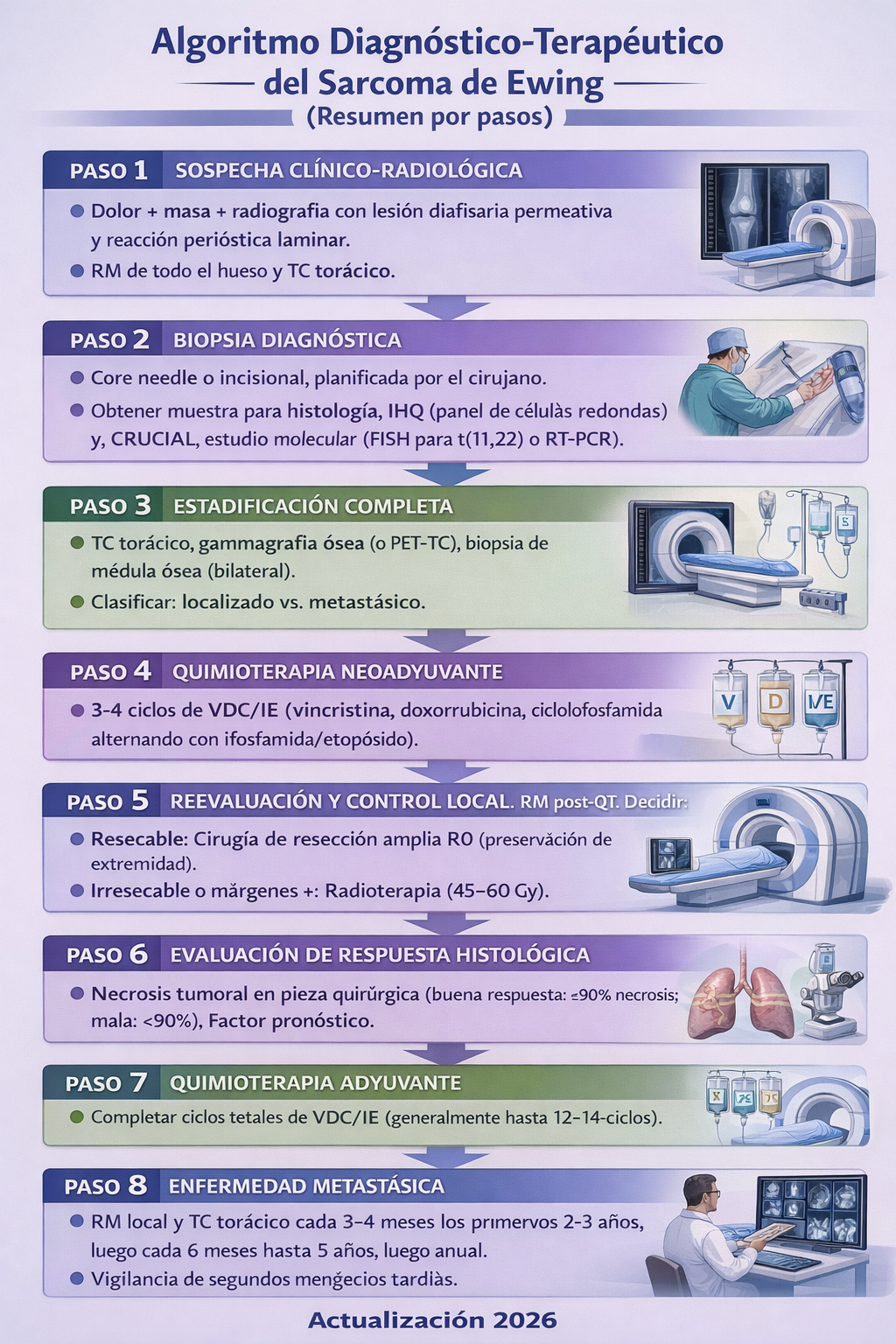
Resumen para la práctica clínica
📌 Indicaciones
- ✅ Confirmación diagnóstica mediante demostración de fusión EWSR1/FUS::ETS (idealmente por RNA-NGS; alternativa FISH/RT-PCR).
- ✅ Quimioterapia sistémica tipo VDC/IE (preferible interval-compressed) en todos los pacientes, seguida de control local (cirugía y/o RT) y completar tratamiento.
- ✅ TC torácico y PET-TC para estadificación; BMAB selectiva según protocolo/riesgo (puede omitirse en localizados con PET-TC negativo en algunos consensos).
- ✅ WLI (15–18 Gy según edad/protocolo) en metástasis pulmonares con respuesta, con boost si residuo macroscópico.
🔧 Técnica
- 🔧 Biopsia: por equipo especializado, con trayecto excindible, asegurando material para molecular.
- 🔧 Quimioterapia: centros con experiencia y soporte hematológico (G-CSF).
- 🔧 Radioterapia: planificación conformada (IMRT/protones cuando proceda) para minimizar toxicidad.
⚠️ Riesgos
- ⚠️ Mala respuesta a QT y enfermedad metastásica al diagnóstico (principal determinante pronóstico).
- ⚠️ Toxicidad por QT: cardiotoxicidad (antraciclinas), nefrotoxicidad (ifosfamida), mielosupresión, infertilidad.
- ⚠️ Toxicidad por RT: crecimiento, fibrosis, segundas neoplasias (riesgo acumulativo).
- ⚠️ Recidiva local o a distancia.
✅ Resultados
- ✅ Enfermedad localizada: SG 5 años típicamente ~70–80% con tratamiento moderno.
- ✅ Metastásica: peor; mejor si metástasis exclusivamente pulmonares que si óseas/médula.
14) Bibliografía
Referencias clave (OMS/WHO, clasificación y guías)
- 📄 WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. IARC; 2020.
- 📄 Strauss SJ, et al. Bone sarcomas: ESMO–EURACAN–GENTURIS–ERN PaedCan CPG. Ann Oncol. 2021;32:1520–1536.
- 📄 Mata Fernández C, et al. Clinical practice guidelines for the treatment of Ewing sarcoma. Clin Transl Oncol. 2024/2025.
- 📄 NCI. Ewing Sarcoma Treatment (PDQ®). Actualización 2024.
Primera línea (quimioterapia estándar)
- 📄 Womer RB, et al. Interval-compressed chemotherapy for localized Ewing sarcoma (COG AEWS0031). J Clin Oncol. 2012;30:4148–4154.
- 📄 Brennan B, et al. EURO-EWING 2012: comparación de regímenes (VIDE vs VDC/IE). Lancet. 2022.
Diagnóstico molecular e IHC
- 📄 Delattre O, et al. EWS-ETS fusion (EWSR1-FLI1). Nature. 1992.
- 📄 Yoshida A, et al. NKX2.2 como marcador útil en sarcoma de Ewing. Am J Surg Pathol. 2012.
- 📄 Dehner CA, et al. Updates on WHO classification for small round cell tumors. Adv Anat Pathol/Semin Diagn Pathol. 2024.
Recaída / terapias emergentes
- 📄 Attia S, et al. Regorafenib en sarcoma de Ewing avanzado (SARC024). Cancer Med. 2022/2023.
- 📄 Italiano A, et al. Cabozantinib (CABONE) en Ewing avanzado. Lancet Oncol. 2020.
- 📄 Meyers PA, et al. TK216 (antagonista EWS::FLI1) fase I/II. J Clin Oncol. 2024.