🦴 Osteocondroma — Actualización 2026: lesión exofítica con continuidad corticomedular, forma solitaria y hereditaria, y criterios de sospecha de transformación maligna
El osteocondroma es la lesión ósea benigna más frecuente. En realidad, en la mayoría de casos se comporta como una alteración del desarrollo (relacionada con la placa de crecimiento) más que como una neoplasia “de novo”. Consiste en una excrecencia ósea recubierta por un capuchón de cartílago, que crece durante la infancia y adolescencia y suele estabilizarse tras el cierre fisario. Puede presentarse como una lesión solitaria (la forma más habitual) o como parte de la exostosis múltiple hereditaria (EMH), también denominada “múltiples osteocondromas”, asociada a variantes patogénicas en EXT1/EXT2. Su importancia clínica se centra en: síntomas mecánicos o compresivos (tendones, bursas, nervios o vasos) y el riesgo poco frecuente de transformación maligna, habitualmente hacia condrosarcoma secundario, mayor en la EMH.
0) En 1 minuto
- ✅ Qué es: excrecencia ósea benigna con capuchón cartilaginoso, típica de metáfisis. Suele detenerse tras el cierre fisario.
- ✅ Dato radiológico clave: continuidad de cortical y cavidad medular con el hueso huésped.
- ✅ Formas:
- 🔹 Solitario: lo más frecuente.
- 🔹 EMH (exostosis múltiple hereditaria): múltiples lesiones, deformidades y mayor riesgo de transformación.
- ✅ Cuándo da síntomas: dolor por roce/bursitis, limitación funcional, compresión nerviosa (parestesias), compresión vascular (raro), fractura del tallo (raro).
- ✅ Transformación maligna (sospecha): dolor nuevo, crecimiento tras madurez esquelética, masa de partes blandas o capuchón cartilaginoso engrosado en imagen.
- ✅ Tratamiento: observación si asintomático y típico; resección marginal completa si hay síntomas, complicaciones o sospecha de transformación.
Siglas y abreviaturas
- 📄 EMH: exostosis múltiple hereditaria (“múltiples osteocondromas”).
- 📄 RM: resonancia magnética.
- 📄 TC: tomografía computarizada.
- 📄 STIR: secuencia de RM con supresión grasa útil para cartílago/edema.
1) Osteocondroma solitario
1.1 Discusión general
El osteocondroma se origina a partir de cartílago fisario ectópico (una “herniación” lateral de la placa de crecimiento), que continúa creciendo mediante osificación endocondral. Por ello, aparece cerca de la metáfisis y suele orientarse alejándose de la articulación.
1.2 Epidemiología
- 📊 Frecuencia: es la lesión ósea benigna más común (aprox. un tercio de los tumores óseos benignos, según series).
- 📊 Edad: diagnóstico habitual en niños, adolescentes y adultos jóvenes; suele estabilizarse tras el cierre fisario.
- 📊 Sexo: ligero predominio masculino.
- 📊 Localización típica: metáfisis de huesos largos, especialmente alrededor de la rodilla (fémur distal y tibia proximal), húmero proximal y pelvis/escápula. En columna es raro, pero puede causar síntomas neurológicos.
1.3 Clínica
- 🩺 Muchos casos son hallazgo incidental.
- 🩺 Masa palpable adherida al hueso, generalmente indolora.
- 🩺 Cuando es sintomático:
- 🔹 Dolor mecánico por roce tendinoso o bursitis (puede simular masa de partes blandas).
- 🔹 Limitación funcional si interfiere con el movimiento o el tendón.
- 🔹 Compresión nerviosa: parestesias o debilidad (según trayecto nervioso).
- 🔹 Compresión vascular (rara): trombosis o seudoaneurisma.
- 🔹 Fractura del tallo (rara), especialmente en pediculados expuestos a trauma.
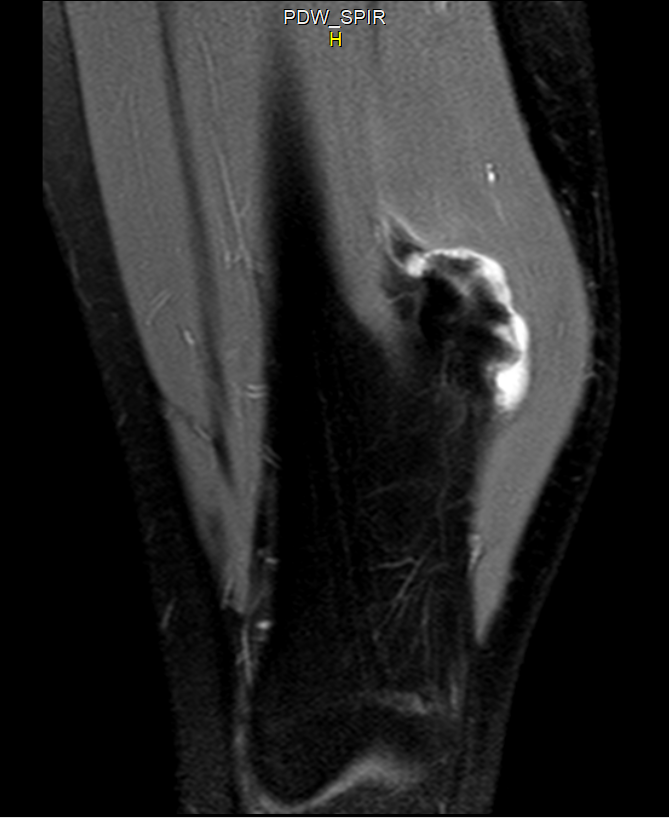
1.4 Imagen
1.4.1 Radiografía simple
- 🩻 Lesión exofítica sésil (base ancha) o pediculada (con tallo).
- 🩻 Signo principal: continuidad corticomedular con el hueso huésped (hallazgo prácticamente diagnóstico).
- 🩻 El capuchón cartilaginoso suele no verse; puede haber calcificaciones irregulares si es grueso o si existe degeneración.
1.4.2 TC y RM
- 🩻 TC: útil en localizaciones complejas (pelvis/escápula/columna) y para definir continuidad corticomedular.
- 🩻 RM: clave para medir el capuchón cartilaginoso (hiperintenso en T2/STIR) y valorar bursitis/compresión neurovascular. Es la prueba más útil ante sospecha de transformación.
Hallazgo radiológico característico
1.5 Histopatología
- 🔬 Capuchón cartilaginoso: cartílago hialino con organización similar a placa de crecimiento; suele ser más grueso en niños/adolescentes y adelgazarse en adultos.
- 🔬 Osificación endocondral: debajo del capuchón, con formación de hueso esponjoso.
- 🔬 Continuidad medular: el hueso trabecular del osteocondroma se continúa con la médula del hueso huésped.
- 🔬 En pacientes inmaduros puede haber hipercelularidad leve sin que implique malignidad (interpretación contextual).
1.6 Genética molecular
- 🧬 En osteocondroma solitario pueden existir alteraciones somáticas relacionadas con EXT1/EXT2, pero sin patrón hereditario.
- 🧬 En EMH, las variantes patogénicas germinales en EXT1 o EXT2 reducen la síntesis de heparán sulfato y alteran señales de crecimiento en la placa fisaria.
1.7 Transformación maligna
Riesgo: bajo en solitario (habitualmente <1%); mayor en EMH (aprox. 5–10% en muchas series, con rangos más amplios según cohortes y criterios).
Claves de sospecha
- ⚠️ Dolor de nueva aparición o claramente progresivo sin causa mecánica evidente.
- ⚠️ Crecimiento después de la madurez esquelética.
- ⚠️ Capuchón cartilaginoso engrosado en RM:
- 🔹 En adultos, un capuchón >2 cm es muy sospechoso.
- 🔹 En niños/adolescentes puede ser más grueso sin malignidad; la interpretación debe ser clínica y evolutiva.
- ⚠️ Masa de partes blandas adyacente o cambios agresivos (destrucción cortical, edema importante no explicable, etc.).
La transformación es casi siempre a condrosarcoma secundario, con frecuencia de bajo grado, especialmente en pelvis y cintura escapular.
1.8 Diagnóstico diferencial
| Entidad | Claves para diferenciar |
|---|---|
| EMH | Múltiples lesiones, antecedentes familiares y deformidades (discrepancia de longitud, deformidad angular). |
| Osteosarcoma parosteal | Masa densa adherida a cortical (frecuente en fémur distal posterior). Suele faltar la continuidad medular típica. Puede asociar amplificación de MDM2 (dato patológico). |
| Condroma periostal | Lesión cartilaginosa yuxtacortical que “muerde” cortical; no hay continuidad corticomedular. |
| Osteoma | Hueso denso bien delimitado, típico de cráneo/senos; sin capuchón cartilaginoso. |
| Displasia epifisaria hemimélica (Trevor) | Origen epifisario (no metafisario), crecimiento unilateral y periarticular. |
El diferencial más relevante en la práctica es distinguir osteocondroma benigno de condrosarcoma secundario; el contexto (edad, crecimiento, dolor) y la medición del capuchón en RM son los datos más útiles.
1.9 Tratamiento
- 🛠️ Observación: si es típico y asintomático.
- 🛠️ Resección quirúrgica (marginal): indicada ante síntomas, complicaciones (bursitis persistente, compresión) o sospecha de transformación.
- 🛠️ Técnica: extirpación completa incluyendo capuchón y pericondrio; osteotomía a nivel de base sobre hueso sano.
- 🛠️ Evolución: muy buena; la recidiva suele relacionarse con resección incompleta.
11) Exostosis múltiple hereditaria (EMH)
2.1 Concepto y genética
- 🧬 Trastorno autosómico dominante con múltiples osteocondromas.
- 🧬 También se denomina “múltiples osteocondromas” o “osteocondromatosis familiar”.
- 🧬 Genética: variantes patogénicas germinales en EXT1 (8q24) o EXT2 (11p11). En general, EXT1 se asocia a fenotipos más graves.
2.2 Clínica
- 🩺 Inicio habitual en la infancia; las lesiones se evidencian conforme avanza el crecimiento.
- 🩺 Mayor frecuencia de:
- 🔹 deformidades (genu valgo, discrepancia de longitud, deformidad del antebrazo),
- 🔹 limitación funcional,
- 🔹 síntomas compresivos.
2.3 Seguimiento práctico
- 📅 Revisión clínica periódica (dolor nuevo, crecimiento, cambios funcionales).
- 📅 Considerar RM dirigida ante síntomas o en localizaciones de mayor riesgo (pelvis, escápula) si hay dudas.
- 📅 Tratamiento de deformidades: puede requerir cirugía correctora (p. ej., osteotomías) además de resecciones selectivas.
2.4 Transformación maligna
- ⚠️ Riesgo mayor que en la forma solitaria.
- ⚠️ Los criterios de sospecha son los mismos (dolor nuevo, crecimiento en adulto, capuchón engrosado, masa de partes blandas).
12) Displasia epifisaria hemimélica (enfermedad de Trevor)
- 🦶 Qué es: trastorno del desarrollo no hereditario con sobrecrecimiento osteocondral unilateral de una o más epífisis (periarticular).
- 🦶 Localización: predomina en miembro inferior (tobillo/astrágalo, fémur distal, tibia distal).
- 🦶 Clínica: deformidad periarticular, limitación y dolor.
- 🦶 Tratamiento: resección si es sintomático o deformante; la histología puede parecerse a osteocondroma, pero la localización (epífisis) es la clave.
13) Algoritmo de manejo

📌 Resumen para la práctica clínica
Indicaciones
- ✅ Radiografía simple en dos proyecciones como prueba inicial.
- ✅ RM si hay síntomas, localización compleja o sospecha de transformación (capuchón cartilaginoso).
- ✅ Observación en lesiones típicas asintomáticas.
- ✅ Resección marginal completa si hay síntomas, complicaciones o criterios de alarma.
- ✅ Seguimiento clínico a largo plazo en EMH (dolor nuevo o crecimiento en adulto = reevaluación).
Técnica
- 🔧 RM: secuencias T1, T2 y STIR; medir capuchón en el punto de mayor grosor.
- 🔧 Cirugía: resección marginal incluyendo capuchón y pericondrio; osteotomía en la base sobre hueso sano.
Riesgos
- ⚠️ No reevaluar un osteocondroma en adulto con dolor nuevo o crecimiento.
- ⚠️ Resección incompleta (capuchón/pericondrio) con recidiva.
- ⚠️ Confundir osteosarcoma parosteal con osteocondroma (diferencial crítico).
- ⚠️ En EMH, infravalorar lesiones pélvicas/escapulares con riesgo mayor de transformación.
Resultados
- ✅ Recidiva tras resección completa: baja y generalmente ligada a resección incompleta.
- ✅ Transformación maligna: rara en solitario; más frecuente en EMH.
14) Bibliografía
📘 Clasificación
- 📄 WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon: IARC Press; 2020. (Osteochondroma / Multiple osteochondromas).
📑 Exostosis múltiple hereditaria
- 📄 Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation update and description of the EXT genes. Hum Mutat. 2024;45(2):123-135. doi:10.1002/humu.24789
- 📄 Pannier S, Legeai-Mallet L. Hereditary multiple exostoses: from genetics to clinical practice. Bone. 2025;182:116789. doi:10.1016/j.bone.2025.116789
⚠️ Transformación maligna
- 📄 Ahmed AR, Tan TS, Unni KK, et al. Secondary chondrosarcoma in osteochondroma: a study of 107 patients. J Bone Joint Surg Am. 2023;105(10):789-798. doi:10.2106/JBJS.22.00891