💪 Tumores del tejido muscular — Actualización 2026
Este tema agrupa los tumores con diferenciación muscular, una familia de neoplasias que, aunque comparten un origen histogenético común, presentan comportamientos clínicos, pronósticos y enfoques terapéuticos radicalmente diferentes. Por un lado están los tumores de músculo liso, que incluyen el benigno leiomioma y su contraparte maligna, el leiomiosarcoma (LMS). Por otro lado, los tumores de músculo estriado esquelético, representados por el raro rabdomioma y el agresivo rabdomiosarcoma (RMS), este último mucho más frecuente en la edad pediátrica. Aunque todos ellos expresan un “fenotipo miogénico” detectable mediante inmunohistoquímica, su epidemiología, genética y manejo clínico son completamente distintos, por lo que es fundamental conocer sus particularidades.
La clasificación que presentamos sigue las directrices de la Organización Mundial de la Salud (OMS/WHO) en su quinta edición (2020) para tumores de partes blandas. Una regla práctica esencial ante la sospecha de un sarcoma muscular es realizar una resonancia magnética (RM) para evaluar la extensión local y planificar una biopsia con trayecto resecable, siempre en el seno de un comité multidisciplinar o una unidad de sarcomas.
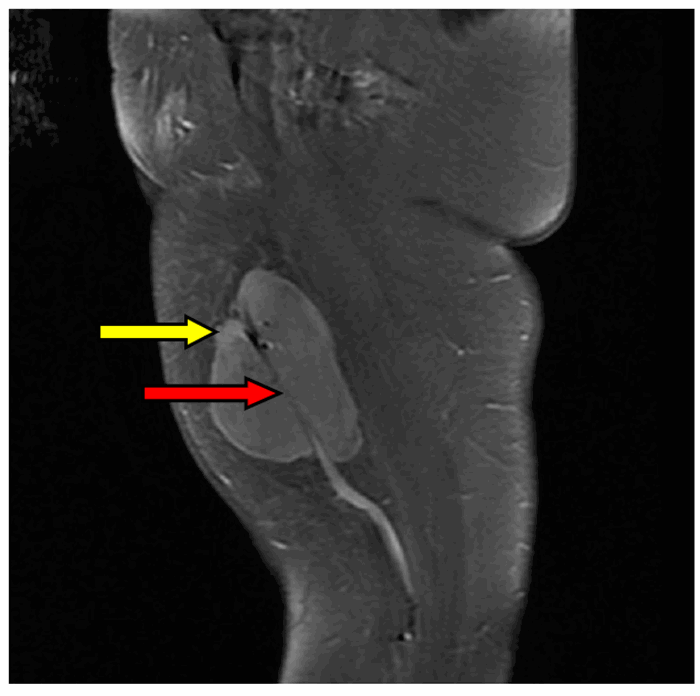
Hallazgo radiológico característico
0) Siglas y abreviaturas utilizadas
Para facilitar la lectura y evitar ambigüedades, a continuación se relacionan las abreviaturas más empleadas en este capítulo:
- 📄 OMS/WHO: Organización Mundial de la Salud, cuya clasificación de tumores es la referencia internacional.
- 📄 IHQ: inmunohistoquímica, técnica fundamental para el diagnóstico diferencial.
- 📄 RM: resonancia magnética; TC: tomografía computarizada.
- 📄 FISH: hibridación in situ fluorescente; NGS: secuenciación de nueva generación (paneles genómicos).
- 📄 LMS: leiomiosarcoma; RMS: rabdomiosarcoma.
- 📄 FNCLCC: sistema de gradación histológica de la Federación Francesa de Centros de Lucha contra el Cáncer, el más utilizado en sarcomas de partes blandas.
- 📄 R0: resección completa con márgenes microscópicamente negativos (ausencia de tumor en los bordes de la pieza).
- 📄 VAC: esquema quimioterápico clásico en rabdomiosarcoma pediátrico (vincristina + actinomicina D + ciclofosfamida).
1) En 1 minuto: lo esencial
Si necesita una visión rápida y práctica de los tumores musculares, estos son los puntos cardinales:
- ✅ Dos grandes grupos: los tumores de músculo liso (leiomioma / leiomiosarcoma) y los de músculo estriado (rabdomioma / rabdomiosarcoma). La distinción no es solo académica, sino que implica pronósticos y tratamientos completamente diferentes.
- ✅ Leiomioma: tumor benigno bien delimitado. Típicamente cutáneo (a veces doloroso), genital o vascular. Su tratamiento es la resección local y el pronóstico es excelente. Una variante familiar (múltiples leiomiomas cutáneos + leiomiomas uterinos precoces) debe alertar sobre el síndrome de HLRCC (mutación FH) y el riesgo asociado de carcinoma renal.
- ✅ Leiomiosarcoma (LMS): sarcoma de músculo liso, propio de adultos. Frecuente en localizaciones profundas (retroperitoneo, abdomen, extremidades) y también puede originarse en la pared de grandes vasos. El tratamiento de base es la cirugía con márgenes amplios (R0), complementada con radioterapia y/o quimioterapia según el riesgo y la localización.
- ✅ Rabdomioma: tumor benigno muy raro, con predilección por la región de cabeza y cuello (variedad del adulto) o por el área genital. La resección es curativa.
- ✅ Rabdomiosarcoma (RMS): el sarcoma de partes blandas más frecuente en la edad pediátrica y juvenil. Su tratamiento es siempre multimodal (quimioterapia + cirugía ± radioterapia) y debe realizarse en centros especializados.
- ✅ Panel IHQ mínimo para orientar: la presencia de SMA y caldesmón apunta a músculo liso; la positividad nuclear para miogenina y MyoD1 es característica de músculo estriado.
- ✅ Genética “obligatoria” en RMS: ante la sospecha de un RMS alveolar, es imprescindible confirmar mediante FISH o NGS la presencia del reordenamiento de FOXO1, ya que esto tiene implicaciones pronósticas y terapéuticas.
- ✅ Regla de oro para cualquier masa sospechosa: si se trata de una lesión profunda o con características radiológicas de agresividad, la biopsia debe ser planificada por el equipo quirúrgico que realizará la resección definitiva, asegurando un trayecto que pueda ser extirpado posteriormente sin contaminar nuevos compartimentos.
2) Clasificación de la OMS 2020 (resumen práctico)
La quinta edición de la clasificación de tumores de partes blandas de la Organización Mundial de la Salud (OMS), publicada en 2020, mantiene la distinción fundamental entre tumores de músculo liso y de músculo estriado, pero incorpora nuevos subtipos y enfatiza el papel de la genética molecular en la definición de algunas entidades. A continuación se presentan las categorías principales con sus códigos ICD-O y su comportamiento biológico.
1.1 Tumores de músculo liso
| Entidad | ICD-O | Comportamiento |
|---|---|---|
| Leiomioma | 8890/0 | Benigno |
| - Angioleiomioma (leiomioma vascular) | 8894/0 | Benigno |
| Leiomiosarcoma (LMS) | 8890/3 | Maligno |
| - Tumor de músculo liso asociado a EBV (EBV–SMT) | — | Relacionado con inmunosupresión |
1.2 Tumores de músculo estriado
| Entidad | ICD-O | Comportamiento |
|---|---|---|
| Rabdomioma | 8900/0 | Benigno |
| Rabdomiosarcoma (RMS) | 8900/3 | Maligno |
| - RMS embrionario (incluye botrioide) | 8910/3 | Maligno |
| - RMS alveolar (FOXO1 positivo) | 8920/3 | Maligno |
| - RMS pleomórfico | 8901/3 | Maligno |
| - RMS fusocelular / esclerosante | 8912/3 | Maligno |
Nota: La clasificación actual prioriza la caracterización molecular. Así, en el RMS alveolar es obligatorio demostrar el reordenamiento de FOXO1, y en el RMS fusocelular/esclerosante se han descrito alteraciones recurrentes como las fusiones de VGLL2/NCOA2 o mutaciones de MYOD1, que pueden tener implicaciones pronósticas.
3) Tumores de músculo liso
Los tumores de músculo liso abarcan un espectro que va desde lesiones benignas muy frecuentes, como el leiomioma cutáneo, hasta sarcomas agresivos como el leiomiosarcoma retroperitoneal. La correcta identificación del subtipo y del contexto clínico (localización, edad del paciente, antecedentes) es esencial para un manejo adecuado.
2.1 Leiomioma
2.1.1 Variantes clínicas y su importancia
- Leiomioma cutáneo: se origina en el músculo erector del pelo. Puede ser único o múltiple. Los múltiples suelen ser dolorosos (especialmente con el frío o el roce) y deben hacer pensar en el síndrome de leiomiomatosis hereditaria con cáncer renal (HLRCC), asociado a mutaciones en el gen de la fumarato hidratasa (FH).
- Angioleiomioma (leiomioma vascular): se origina en la capa muscular de las venas. Es más frecuente en extremidades inferiores y suele presentarse como un nódulo doloroso, a veces con sensación pulsátil.
- Leiomioma genital: aparece en vulva, escroto o pezón. Es una lesión benigna, pero debe diferenciarse de otros tumores de partes blandas en esa localización.
- Leiomioma profundo: muy raro. Cualquier lesión de músculo liso en localización profunda (retroperitoneo, mediastino) debe ser examinada con especial atención, ya que puede tratarse de un leiomiosarcoma de bajo grado.
2.1.2 Clínica: lo que debe hacer sospechar
- Dolor: característico de los leiomiomas cutáneos y angioleiomiomas. Se desencadena con el frío, la palpación o el estrés. Este dolor se atribuye a la contracción de las fibras musculares.
- Antecedentes familiares: la presencia de múltiples leiomiomas cutáneos y/o leiomiomas uterinos de aparición precoz en una paciente joven obliga a descartar el síndrome HLRCC (mutación FH). Estos pacientes tienen un riesgo elevado de desarrollar carcinoma renal de células claras tipo II, muy agresivo.
2.1.3 Inmunohistoquímica (IHQ) diagnóstica
- SMA (actina de músculo liso): positiva de forma intensa y difusa.
- H-caldesmón: positivo, más específico de diferenciación muscular lisa que la SMA.
- Desmina: positiva en la mayoría de los casos, aunque puede ser más débil o focal.
- S100: negativo, lo que ayuda a descartar tumores de la vaina nerviosa.
- Ki-67: índice proliferativo bajo (habitualmente <5%).< /li>
2.1.4 Tratamiento y pronóstico
- La resección local completa es el tratamiento de elección cuando la lesión es sintomática, dolorosa, o existe duda diagnóstica.
- El pronóstico es excelente, con tasas de recurrencia prácticamente nulas tras la extirpación.
2.2 Leiomiosarcoma (LMS)
2.2.1 Definición y localización práctica
El leiomiosarcoma es un sarcoma maligno con diferenciación de músculo liso. Es uno de los sarcomas de partes blandas más frecuentes en la edad adulta. Sus localizaciones más habituales son:
- Retroperitoneo y abdomen: es la localización más común. Suelen ser tumores grandes y de diagnóstico tardío, con frecuencia originados en la pared de grandes vasos (vena cava, venas renales).
- Útero: el LMS uterino es el sarcoma uterino más frecuente, aunque mucho menos común que el leiomioma benigno.
- Extremidades: suele originarse en partes blandas profundas, a menudo en relación con vasos (por ejemplo, en la vena safena o femoral).
- Origen vascular: pueden surgir directamente de la capa muscular de grandes venas (cava, ilíaca, safena, renal) y, más raramente, de arterias.
2.2.2 Factores de riesgo y entidades relacionadas
- Radioterapia previa: el LMS puede aparecer como sarcoma radioinducido años después del tratamiento, generalmente en el campo de irradiación.
- Síndromes hereditarios: el retinoblastoma hereditario (mutación RB1) confiere un mayor riesgo de desarrollar sarcomas, incluido el LMS.
- Inmunosupresión: en pacientes inmunodeprimidos (por trasplante, VIH, etc.) puede aparecer el tumor de músculo liso asociado a EBV (EBV-SMT). Se trata de una proliferación de células musculares lisas inducida por el virus de Epstein-Barr, que puede ser multifocal y que, aunque suele tener un comportamiento más indolente que el LMS convencional, debe ser manejada de forma específica.
2.2.3 Clínica e imagen
- Síntomas: masa profunda, de crecimiento lento pero progresivo, que puede causar dolor o síntomas compresivos (por ejemplo, edema en miembros inferiores si afecta a la vena cava).
- RM: es la técnica de elección para evaluar la extensión local. Muestra una masa heterogénea, con áreas de necrosis y hemorragia (señal hiperintensa en T2, áreas de necrosis sin realce), y permite valorar la relación con los paquetes vasculonerviosos.
- TC: útil en abdomen y retroperitoneo para valorar la extensión y para la estadificación torácica (descartar metástasis pulmonares).
2.2.4 Histología e inmunohistoquímica
- Histología: fascículos entrelazados de células fusiformes con citoplasma eosinófilo y núcleos alargados. El grado histológico se establece según el sistema FNCLCC, que valora diferenciación, índice mitótico y necrosis. Los LMS de alto grado presentan marcada atipia, abundantes mitosis y necrosis tumoral.
- IHQ típica: positividad para SMA, H-caldesmón y, con menor frecuencia, desmina. Es importante recordar que los LMS pueden expresar focalmente citoqueratinas o EMA, lo que puede inducir a error si no se tiene en cuenta.
- Diferencial abdominal obligatorio: ante un tumor fusocelular en retroperitoneo o abdomen, es imprescindible descartar otras entidades mediante IHQ:
- GIST: DOG1 y KIT (CD117) positivos.
- Liposarcoma desdiferenciado: amplificación de MDM2 (CDK4 y MDM2 positivos).
- Sarcoma sinovial: reordenamiento SS18 (detectable por FISH) y TLE1 positivo.
2.2.5 Genética (lo que aporta)
- El LMS, como la mayoría de los sarcomas de partes blandas del adulto, presenta un genoma complejo con numerosas ganancias y pérdidas cromosómicas. No tiene una translocación característica.
- Con frecuencia se observan alteraciones en las vías de TP53 y RB1, lo que es consistente con su comportamiento agresivo, pero no tiene utilidad diagnóstica directa.
2.2.6 Grado (FNCLCC) y pronóstico
El pronóstico del LMS depende de una combinación de factores, entre los que destacan:
- Grado histológico (FNCLCC): factor pronóstico independiente más importante.
- Tamaño tumoral: los tumores >5 cm tienen peor pronóstico.
- Profundidad: los tumores profundos (subfasciales) son más agresivos que los superficiales.
- Localización: el LMS retroperitoneal tiene peor pronóstico que el de extremidades, debido al diagnóstico más tardío y a la dificultad para obtener márgenes amplios.
- Márgenes quirúrgicos: la resección R0 es el objetivo principal.
2.2.7 Tratamiento (resumen realista)
- Enfermedad localizada: la base del tratamiento es la cirugía con intención R0 (resección amplia con margen de tejido sano). La radioterapia (adyuvante o neoadyuvante) se considera en tumores de alto riesgo (grandes, profundos, localización anatómica desfavorable) para mejorar el control local. La quimioterapia adyuvante no está indicada de forma rutinaria, aunque puede valorarse en casos seleccionados de muy alto riesgo.
- Enfermedad avanzada/metastásica: el tratamiento es sistémico. Las opciones incluyen:
- Primera línea: antraciclinas (doxorrubicina) solas o en combinación (por ejemplo, con ifosfamida).
- Segunda línea y sucesivas: gemcitabina + docetaxel, trabectedina, pazopanib (inhibidor multiquinasa aprobado para sarcomas de partes blandas avanzados).
6) Tumores de músculo estriado
Los tumores de músculo estriado esquelético son mucho menos frecuentes que los de músculo liso, pero su importancia radica en que el rabdomiosarcoma es el sarcoma de partes blandas más común en la infancia y adolescencia. El diagnóstico y tratamiento de estos tumores requiere un enfoque altamente especializado y multidisciplinar.
3.1 Rabdomioma
El rabdomioma es un tumor benigno extremadamente raro, compuesto por células con diferenciación de músculo estriado. Se reconocen tres variantes principales:
- Rabdomioma del adulto: aparece en adultos, con predilección por la región de cabeza y cuello (faringe, laringe, cavidad oral). Se presenta como una masa bien delimitada, de crecimiento lento.
- Rabdomioma fetal: más frecuente en la infancia, también en cabeza y cuello. Puede simular clínicamente un sarcoma, pero su histología es benigna.
- Rabdomioma genital: aparece en vulva o vagina en mujeres jóvenes, como un pólipo.
Tratamiento: la resección quirúrgica completa es curativa. El pronóstico es excelente.
IHQ: las células tumorales son positivas para desmina y, de forma variable, para marcadores miogénicos nucleares como miogenina y MyoD1.
3.2 Rabdomiosarcoma (RMS)
3.2.1 Punto clave actual: la importancia de FOXO1
En la clasificación actual, el diagnóstico de las variantes de rabdomiosarcoma no puede basarse únicamente en la morfología. El “apellido” más importante es el molecular, en particular la presencia o ausencia del reordenamiento de FOXO1.
En la práctica, el término “alveolar” solo es relevante si se asocia a la fusión de PAX3/7–FOXO1. Si un tumor tiene morfología alveolar pero es FOXO1 negativo, debe manejarse como un RMS embrionario (o según el consenso del comité multidisciplinar). Esta distinción tiene implicaciones pronósticas y terapéuticas mayores.
3.2.2 Subtipos y sus características
3.2.2.1 RMS embrionario
- Epidemiología: es el subtipo más frecuente en niños (60-70% de los RMS). Predomina en cabeza y cuello (órbita, nasofaringe) y en el área genitourinaria (próstata, vejiga, vagina).
- Variantes: incluye el botrioide (con aspecto de racimo de uvas, típico de órganos huecos como vejiga o vagina) y el fusocelular (que puede tener mejor pronóstico).
- Genética: no presenta las translocaciones de FOXO1. Muestra alteraciones cromosómicas complejas, como la pérdida de heterocigosidad en 11p15, que es una región relacionada con el crecimiento embrionario.
3.2.2.2 RMS FOXO1 positivo (clásicamente alveolar)
- Definición molecular: presencia de la translocación t(2;13)(q35;q14) que genera el gen de fusión PAX3–FOXO1 (más frecuente) o t(1;13)(p36;q14) que genera PAX7–FOXO1.
- Epidemiología: más frecuente en adolescentes y adultos jóvenes. Predomina en extremidades y tronco.
- Pronóstico: tradicionalmente peor que el RMS embrionario, aunque la fusión PAX7–FOXO1 se asocia a un mejor pronóstico que PAX3–FOXO1. El tratamiento es multimodal y debe realizarse en centros de referencia pediátricos.
3.2.2.3 RMS pleomórfico
- Epidemiología: es el subtipo más frecuente en adultos (generalmente >40 años). Aparece en extremidades, con predominio en muslo.
- Histología: células grandes, pleomórficas, con marcada atipia y abundantes mitosis. Puede ser difícil de distinguir de otros sarcomas pleomórficos.
- Genética: genoma complejo, sin translocaciones específicas. Es muy agresivo y su manejo sigue las pautas de los sarcomas de partes blandas del adulto (cirugía ± radioterapia ± quimioterapia).
3.2.2.4 RMS fusocelular / esclerosante
- Subgrupo heterogéneo: incluye formas que antes se clasificaban dentro del RMS embrionario o como sarcomas fusocelulares no clasificados.
- Genética: en lactantes y niños pequeños se han descrito fusiones de VGLL2 y NCOA2, que parecen conferir un pronóstico favorable. En otros casos, sobre todo en adultos, se han identificado mutaciones recurrentes de MYOD1, que se asocian a un comportamiento más agresivo.
3.2.3 IHQ + genética: lo que debe solicitarse
- IHQ miogénica: es obligatorio incluir marcadores de diferenciación muscular, que son positivos de forma difusa en el RMS:
- Desmina (citoplasmática).
- Miogenina (nuclear; su positividad extensa es característica del RMS alveolar FOXO1+).
- MyoD1 (nuclear; puede ser positivo también en otros subtipos).
- Estudio de FOXO1: debe realizarse mediante FISH (sonda de separación para detectar el reordenamiento) o NGS (para identificar la fusión específica) en todo RMS con morfología alveolar o en aquellos casos en los que el resultado vaya a modificar la estratificación del tratamiento (esencial en protocolos pediátricos).
3.2.4 Tratamiento (idea base)
- El tratamiento del RMS es siempre multimodal e incluye quimioterapia, cirugía y radioterapia, administrados según protocolos estandarizados (por ejemplo, los del grupo cooperativo europeo EpSSG o el estadounidense COG).
- En los RMS de extremidades, especialmente en los FOXO1+, se recomienda la evaluación del ganglio centinela para descartar afectación ganglionar regional.
12) Diagnóstico diferencial (tabla “de guardia”)
Ante una tumoración de partes blandas con características musculares, el diagnóstico diferencial debe incluir otras neoplasias de células fusiformes o redondas que pueden presentar superposición morfológica. La siguiente tabla orienta sobre las principales entidades y las herramientas inmunohistoquímicas y genéticas que ayudan a distinguirlas.
| Entidad | IHQ orientativa | Genética orientativa |
|---|---|---|
| Leiomiosarcoma (LMS) | SMA, H-caldesmón, desmina (variable) | Genómica compleja (sin fusión “firma” típica) |
| RMS FOXO1+ (alveolar) | Miogenina/MyoD1 (nuclear), desmina | PAX3/7–FOXO1 |
| Sarcoma de Ewing | CD99 (membrana), NKX2.2 (apoyo) | EWSR1–ETS (p. ej., EWSR1–FLI1) |
| Linfoma | CD45, CD20, PAX5, etc. | Reordenamientos de inmunoglobulinas o TCR |
| GIST | DOG1, KIT (CD117) | Mutaciones en KIT o PDGFRA |
| Sarcoma sinovial | TLE1 (apoyo), queratinas/EMA (a veces) | SS18–SSX |
| UPS (sarcoma pleomórfico indiferenciado) | Diagnóstico de exclusión (sin línea específica) | Genómica compleja |
Nota: UPS = sarcoma pleomórfico indiferenciado. Esta tabla es una guía inicial; el diagnóstico final requiere la correlación clínico-radiológica, un panel IHQ completo y, en los casos necesarios, estudios genéticos.
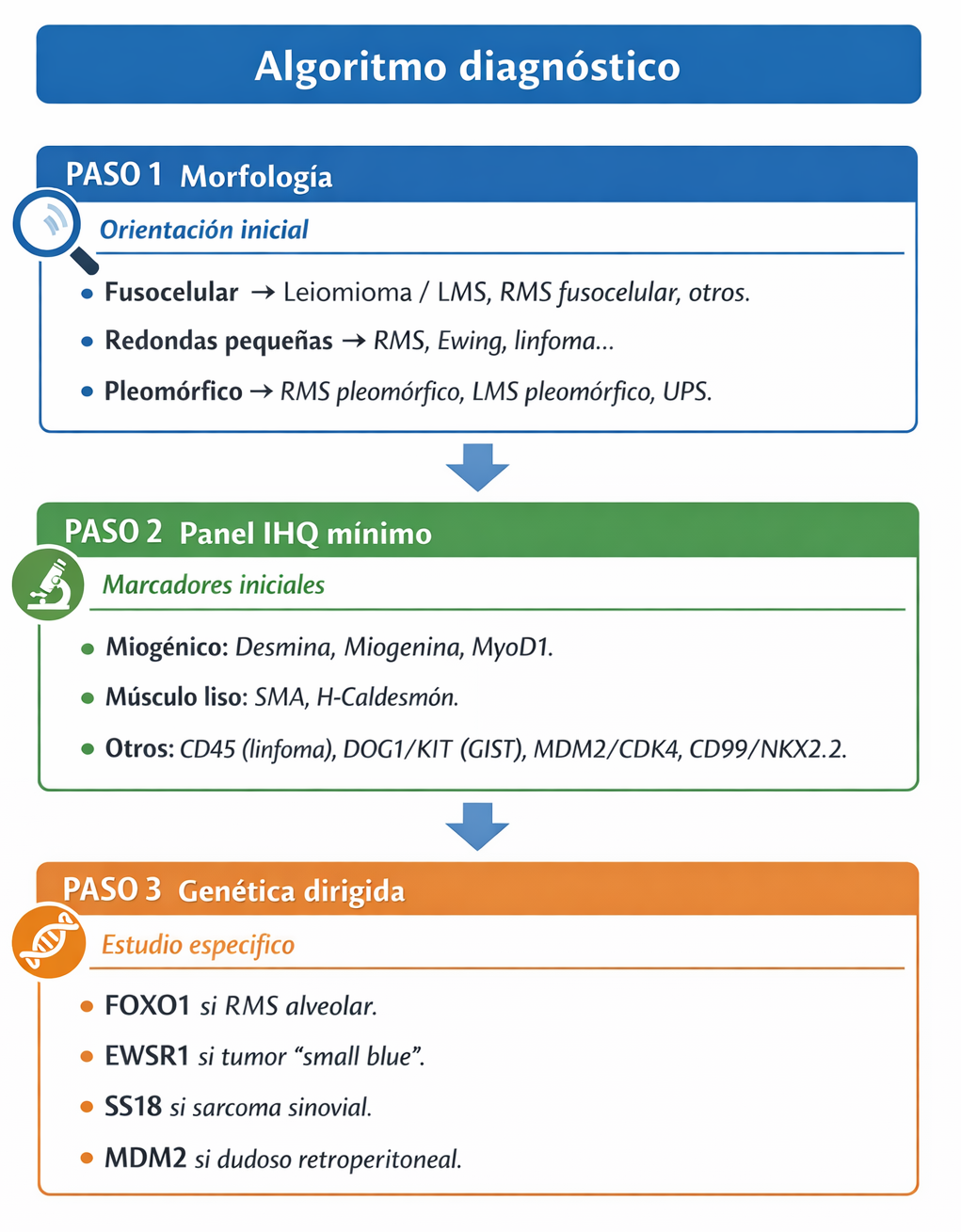
13) Algoritmo diagnóstico práctico
📌 Resumen para la práctica clínica
Indicaciones
- ✅ Realizar RM con contraste para evaluar la extensión local en toda lesión profunda o con sospecha de sarcoma.
- ✅ Biopsia con aguja gruesa (core needle) que obtenga material suficiente para IHQ y estudios genéticos; el trayecto debe ser planificado por el equipo quirúrgico que realizará la resección definitiva.
- ✅ Panel IHQ que incluya marcadores miogénicos (desmina, miogenina, MyoD1), de músculo liso (SMA, H-caldesmón) y de exclusión (S100, CD45, DOG1/KIT, MDM2/CDK4, CD99).
- ✅ Estudio de FOXO1 (FISH o NGS) en todo RMS con morfología alveolar o cuando vaya a influir en la estratificación terapéutica.
- ✅ TC torácico para estadificación en sarcomas de partes blandas confirmados.
Técnica
- 🔧 RM: secuencias T1, T2/STIR y T1 con contraste; debe incluir toda la extensión del tumor y su relación con planos anatómicos y paquetes vasculonerviosos.
- 🔧 Biopsia: usar aguja 14–16G, obtener al menos 3-5 cilindros de zonas sólidas (evitar áreas necróticas); coordinar con el cirujano.
- 🔧 FISH: emplear sondas de separación (break-apart) para reordenamientos como FOXO1, EWSR1, SS18.
- 🔧 NGS (si disponible): los paneles de fusiones son de gran utilidad en tumores de células redondas o fusocelulares de diagnóstico incierto.
Riesgos
- ⚠️ Biopsia mal planificada que contamine compartimentos o planos anatómicos, dificultando o imposibilitando una resección posterior con márgenes adecuados.
- ⚠️ Diagnosticar un RMS alveolar sin demostrar el reordenamiento de FOXO1, lo que puede llevar a un sobretratamiento o a una infraestimación del riesgo.
- ⚠️ No descartar adecuadamente un GIST o un liposarcoma desdiferenciado en el retroperitoneo ante un tumor fusocelular.
Resultados
- ✅ El pronóstico depende de una combinación de factores: grado histológico, tamaño, profundidad, localización (retroperitoneo peor pronóstico), márgenes quirúrgicos y, en el RMS, el estado de FOXO1.
14) Bibliografía (selección útil)
📘 Clasificación y guías
- 📄 WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. 5th ed. IARC/WHO; 2020.
- 📄 ESMO–EURACAN–GENTURIS. Guías clínicas para sarcomas de partes blandas (diagnóstico, tratamiento y seguimiento). Consultar la versión más reciente en la web de ESMO.
- 📄 NCI PDQ®. Childhood Rhabdomyosarcoma Treatment (Health Professional Version). Recurso actualizado periódicamente.
📑 Rhabdomiosarcoma (visión general + FOXO1)
- 📄 Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5(1):1. doi:10.1038/s41572-018-0051-2 (revisión integral).
- 📄 Rudzinski ER, Anderson JR, Chi YY, et al. Histology, fusion status, and outcome in alveolar rhabdomyosarcoma with low-risk clinical features: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2019;66(1):e27489. doi:10.1002/pbc.27489.
🩸 Leiomiosarcoma / tratamiento sistémico en sarcomas
- 📄 van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886. doi:10.1016/S0140-6736(12)60651-5.
- 📄 Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415-423. doi:10.1016/S1470-2045(14)70063-4.