🦠 Sarcoma con diferenciación heteróloga (término histórico: mesenquimoma maligno) — Actualización 2026
El término “mesenquimoma maligno” se utilizó históricamente para describir sarcomas que, en un mismo tumor,
mostraban dos o más líneas de diferenciación mesenquimal (por ejemplo, componentes osteosarcomatosos, condroideos,
rabdomioblásticos o lipogénicos). Con la evolución de la inmunohistoquímica y, sobre todo, de la genética molecular,
la mayoría de los casos “clásicos” han demostrado corresponder a entidades específicas (p. ej., liposarcoma desdiferenciado
con diferenciación heteróloga, sarcoma sinovial, MPNST con componente heterólogo, condrosarcoma mesenquimal, etc.).
Por esa razón, en la práctica moderna y en la terminología OMS 2020, “mesenquimoma maligno” se considera un término histórico
y se evita como diagnóstico principal.
A día de hoy, el enfoque correcto es jerárquico: primero hay que agotar la reclasificación hacia una entidad definida (con su
implicación terapéutica y pronóstica) y, solo si eso no es posible tras un estudio completo y un muestreo adecuado,
utilizar un diagnóstico de exclusión como “sarcoma indiferenciado de alto grado / sarcoma pleomórfico indiferenciado (UPS)
con diferenciación heteróloga”.
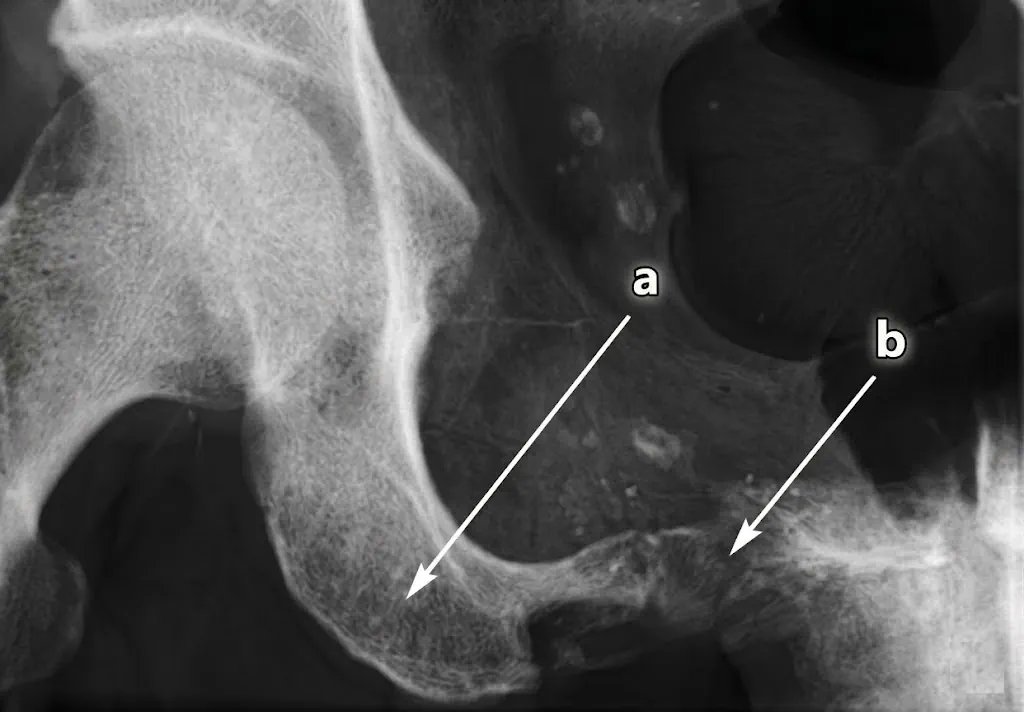
Hallazgo radiológico característico
0) En 1 minuto
- ✅ Idea clave: “Mesenquimoma maligno” es un rótulo histórico. Hoy se debe intentar un diagnóstico más específico; si no es posible, se usa “UPS / sarcoma indiferenciado con diferenciación heteróloga” como diagnóstico de exclusión.
- ✅ Cuándo se plantea: cuando el tumor muestra componentes heterólogos (p. ej., rabdomioblástico, osteosarcomatoso, condroide, lipogénico) y no encaja de forma clara en una entidad definida.
- ✅ Qué no hacer: diagnosticar “mesenquimoma maligno” sin (1) muestreo amplio, (2) panel IHQ orientado y (3) estudio molecular dirigido (al menos para las entidades más probables).
- ✅ Tratamiento: debe basarse en el diagnóstico final. Si se reclasifica (p. ej. Ewing, sarcoma sinovial, etc.), se siguen protocolos específicos. Si queda como sarcoma indiferenciado de alto grado, se trata como sarcoma de alto grado en unidad de referencia.
1) Conceptos
1.1 Qué significaba históricamente “mesenquimoma maligno”
En la literatura clásica, “mesenquimoma maligno” se aplicaba a sarcomas que mostraban dos o más tipos de tejido mesenquimal maligno en la misma neoplasia (por ejemplo, osteosarcoma + liposarcoma; condrosarcoma + rabdomiosarcoma). Era un término descriptivo útil cuando no existían herramientas para afinar el diagnóstico.
1.2 Enfoque actual OMS-compatible (2026)
En la práctica moderna, el camino recomendado es:
- Identificar y documentar los componentes en H&E (y confirmar con IHQ cuando corresponda).
- Buscar una entidad específica con ayuda de marcadores y genética (fusiones, amplificaciones).
- Si tras una evaluación completa no se demuestra una entidad definida, usar un diagnóstico de exclusión como sarcoma indiferenciado de alto grado / UPS con diferenciación heteróloga.
Esto no es un cambio “de nombre”: es un cambio de método. El objetivo es que el diagnóstico final sea útil para decidir tratamiento y pronóstico.
1.3 ICD-O (codificación) vs. “entidad OMS”
Es normal encontrar el código ICD-O 8990/3 (“mesenchymoma, malignant”) en bases de datos. Eso refleja una categoría de codificación histórica, no que la OMS recomiende ese diagnóstico como etiqueta principal.
2) Epidemiología
En hueso es un fenómeno excepcional y la literatura se compone de casos aislados y pequeñas series históricas. Hoy, muchos casos antiguos se reclasificarían a entidades concretas. Por ello, no tiene sentido dar porcentajes “finos”: el mensaje real es que, cuando aparece, debe tratarse como sarcoma de alto grado hasta demostrar lo contrario.
- Edad: variable según el subtipo final (algunos se concentran en jóvenes; otros en adultos).
- Localización: también variable; si es primario óseo, suele presentarse como lesión agresiva con masa de partes blandas.
3) Clínica
La clínica suele ser la de un sarcoma agresivo: dolor progresivo, masa palpable cuando hay extensión a partes blandas, y deterioro funcional local. No hay síntomas sistémicos específicos, pero pueden aparecer por carga tumoral o complicaciones.
- Dolor: habitualmente progresivo.
- Masa / tumefacción: frecuente si hay componente extraóseo.
- Fractura patológica: posible en lesiones óseas extensas.
4) Imagen
En imagen, el patrón suele ser agresivo e inespecífico. El punto clave no es “adivinar” el diagnóstico exacto por imagen, sino estadiar y planificar una biopsia segura.
- RX/TC: lesión lítica agresiva, destrucción cortical, reacción perióstica variable y masa de partes blandas.
- RM: define extensión local, compartimentos, neurovasculares y heterogeneidad interna (útil para dirigir muestreo).
- Estadiaje: TC tórax (pulmón) y, según caso, PET/TC o gammagrafía ósea, en protocolo de sarcomas.
5) Histopatología
5.1 Qué buscar (y por qué)
Lo definitorio es la coexistencia de componentes heterólogos. En lugar de memorizar combinaciones “clásicas”, es más útil pensar en categorías de diferenciación:
| Componente | Pistas morfológicas | Confirmación habitual |
|---|---|---|
| Osteosarcomatoso | Producción de osteoide tumoral por células malignas | SATB2 (apoyo), correlación con mineralización |
| Condroide | Matriz condroide con atipia (según entidad) | S100/SOX9 (apoyo), genética si sospecha mesenquimal |
| Rabdomioblástico | Células eosinófilas, rabdomioblastos | Miogenina, MyoD1 |
| Lipogénico | Lipoblastos, atipia adipocítica | MDM2/CDK4 (si sospecha DDLPS; confirmar amplificación) |
Importante: muchos sarcomas “definidos” pueden tener diferenciación heteróloga (p. ej., DDLPS, MPNST). Por eso, ver 2 componentes no basta para “bautizar” mesenquimoma: hay que reclasificar.
6) Inmunohistoquímica
La IHQ sirve para confirmar líneas de diferenciación y, sobre todo, para orientar el estudio molecular. No reemplaza a la genética cuando se sospechan entidades definidas por fusión/amplificación.
| Objetivo | Marcadores útiles | Comentario |
|---|---|---|
| Rabdomioblástico | Miogenina, MyoD1, desmina | Miogenina/MyoD1 son los más específicos |
| Neural/MPNST | S100/SOX10 (focal), H3K27me3 (pérdida) | La pérdida de H3K27me3 apoya MPNST en el contexto adecuado |
| SFT | STAT6 (nuclear) | Hemangiopericitoma es término histórico; hoy se integra en SFT |
| Ewing/relacionados | CD99, NKX2.2 (apoyo) | Requiere confirmación molecular (EWSR1/FUS) |
| Sinovial | TLE1 (apoyo), EMA/citoqueratinas (variables) | Confirmación por SS18::SSX |
| DDLPS/MDM2 | MDM2, CDK4 (apoyo) | Confirmar amplificación (FISH/NGS); la IHQ sola puede fallar |
7) Genética molecular / reclasificación
En este contexto, la genética es parte del diagnóstico, no un “extra”. El objetivo es contestar una pregunta concreta: ¿puedo asignar una entidad OMS definida?
- MDM2 (amplificación): apoya DDLPS (o ciertos osteosarcomas de bajo grado). Si es positiva, el diagnóstico suele cambiar de forma relevante.
- SS18::SSX: confirma sarcoma sinovial.
- EWSR1/FUS (reordenamientos/fusiones): sitúa el tumor en la familia Ewing/otros tumores EWSR1-reordenados según contexto.
- HEY1::NCOA2: confirma condrosarcoma mesenquimal.
- NGS: útil cuando el panel dirigido es negativo o cuando hay morfología compleja (puede detectar fusiones raras o perfiles compatibles con entidad específica).
8) Diagnóstico diferencial (los “grandes imitadores”)
| Entidad | Por qué se confunde | Clave para separarla |
|---|---|---|
| DDLPS con diferenciación heteróloga | Puede tener osteosarcoma/rabdomiosarcoma/condroide | MDM2 amplificado (confirmación molecular) |
| MPNST con componente heterólogo (tumor Tritón) | Diferenciación rabdomioblástica | Contexto (NF1), patrón, pérdida H3K27me3 (apoyo) |
| Sarcoma sinovial | Heterogeneidad, marcadores variables | SS18::SSX |
| Condrosarcoma mesenquimal | Puede tener patrón bifásico | HEY1::NCOA2 |
| UPS / sarcoma indiferenciado | Cajón de sastre si no se estudia bien | Diagnóstico de exclusión tras panel completo |
9) Tratamiento
9.1 Regla práctica
El tratamiento no se decide por la etiqueta “mesenquimoma maligno”, sino por el diagnóstico final tras reclasificación. Por eso, lo correcto es:
- Si se demuestra una entidad específica: tratar según guías/protocolos del subtipo.
- Si queda como sarcoma indiferenciado de alto grado/UPS con diferenciación heteróloga: tratar como sarcoma de alto grado en unidad de sarcomas.
9.2 Esquema general (orientativo)
- Primario óseo de alto grado (cuando aplica): cirugía con intención R0 + quimioterapia según protocolo del centro (a menudo similar a osteosarcoma/UPS óseo).
- Partes blandas: cirugía ± radioterapia ± quimioterapia según tamaño, profundidad, márgenes y subtipo (guías STS).
- Metástasis pulmonares: valoración individual (cirugía/ablación/RT estereotáxica ± sistémico) en comité.
10) Pronóstico
El pronóstico depende casi por completo de dos factores: (1) el subtipo real tras reclasificación y (2) el estadio (tamaño, márgenes, metástasis). Si el tumor se comporta como un sarcoma de alto grado, el riesgo principal es la metástasis pulmonar, además de la recidiva local si los márgenes son insuficientes.
- Seguimiento: siguiendo esquemas de sarcomas de alto grado (control local + tórax) y adaptado al subtipo.
- Consejo práctico: si el diagnóstico final queda como “UPS/sarcoma indiferenciado”, prioriza segunda opinión en patología experta y NGS si es accesible.
11) Algoritmo diagnóstico (reclasificación primero)

📌 Resumen para la práctica clínica
Indicaciones
- ✅ Sarcoma óseo/partes blandas de alto grado con morfología heterogénea o componentes heterólogos.
- ✅ Necesidad de reclasificación con IHQ + genética (panel dirigido y/o NGS).
- ✅ Manejo en unidad de sarcomas con comité multidisciplinar.
Técnica
- 🔧 Biopsia planificada con trayecto excindible y material para H&E, IHQ y genética (FISH/NGS).
- 🔧 Panel molecular mínimo según sospecha: MDM2, SS18, EWSR1/FUS, HEY1::NCOA2 (y otros según contexto).
- 🔧 Tratamiento guiado por diagnóstico final (subtipo específico o UPS/sarcoma indiferenciado).
Riesgos
- ⚠️ Usar “mesenquimoma maligno” como diagnóstico primario sin excluir entidades específicas.
- ⚠️ Muestreo insuficiente de tumor heterogéneo (diagnóstico incompleto).
- ⚠️ Interpretar IHQ (p. ej. MDM2) sin confirmación de amplificación cuando es relevante.
- ⚠️ Planificar cirugía/biopsia fuera de circuito de sarcomas (contaminación de compartimentos).
Resultados
- ✅ El pronóstico depende del subtipo real y del estadio; si es de alto grado, riesgo de metástasis pulmonar.
- ✅ La reclasificación correcta mejora decisiones terapéuticas y evita tratamientos inadecuados.
12) Bibliografía
📘 Concepto histórico
- 📄 Stout AP. Mesenchymoma, the mixed tumor of mesenchymal derivatives. Ann Surg. 1948.
- 📄 Schajowicz F, Cuevillas AR, Silberman FS. Primary malignant mesenchymoma of bone. J Bone Joint Surg Am. 1966.
- 📄 Kessler S, Mirra JM, Ishii T, Thompson JC, Brien EW. Primary malignant mesenchymoma of bone: case report and literature review. Skeletal Radiol. 1995;24(4):291–295.
📑 Por qué hoy se evita el término (reclasificación)
- 📄 Adachi T, et al. Prognostic factors in the so-called malignant mesenchymoma. Hum Pathol. 2003.
- 📄 Sbaraglia M, Dei Tos AP. (Revisión) El término “malignant mesenchymoma” se abandona y se priorizan diagnósticos específicos / sarcomas con diferenciación heteróloga. Soft Tissue Sarcomas. Cambridge; 2019.
- 📄 Patrichi AI, et al. Pathogenetic and molecular classifications… (menciona que “malignant mesenchymoma” ya no está disponible en la 5ª edición OMS). Pathol Res Pract. 2024.
- 📄 WHO / ICD-O. International Classification of Diseases for Oncology: mantiene 8990/3 (codificación), no como entidad OMS vigente.