🦴 Mesenquimoma fibrocartilaginoso (Fibrocartilaginous mesenchymoma) — Actualización 2026
El mesenquimoma fibrocartilaginoso (fibrocartilaginous mesenchymoma, FCM) es un tumor óseo
extraordinariamente infrecuente descrito inicialmente por Dahlin y colaboradores en 1984.
Desde entonces se han publicado menos de 40 casos (la cifra exacta varía según los criterios diagnósticos de cada revisión).
Su interés práctico no está en la frecuencia, sino en el riesgo de confusión: en imagen puede parecer una lesión fibrosa benigna
(por ejemplo, una displasia fibrosa con cartílago) y, en histología, el componente fusocelular puede inducir a sobrediagnosticar
un sarcoma si la muestra es incompleta.
En la OMS 2020 se incluye dentro de “Other mesenchymal tumours of bone” y se codifica como entidad
de comportamiento intermedio (localmente agresivo) (ICD-O 8990/1, según corrección oficial).
En las series clínicas, el patrón de comportamiento es consistente: tiende a recidivar cuando la resección es incompleta
(curetaje o excisión intralesional), mientras que la metástasis no es un rasgo típico (no se han documentado metástasis
confirmadas en las series de referencia; el problema real es el control local).
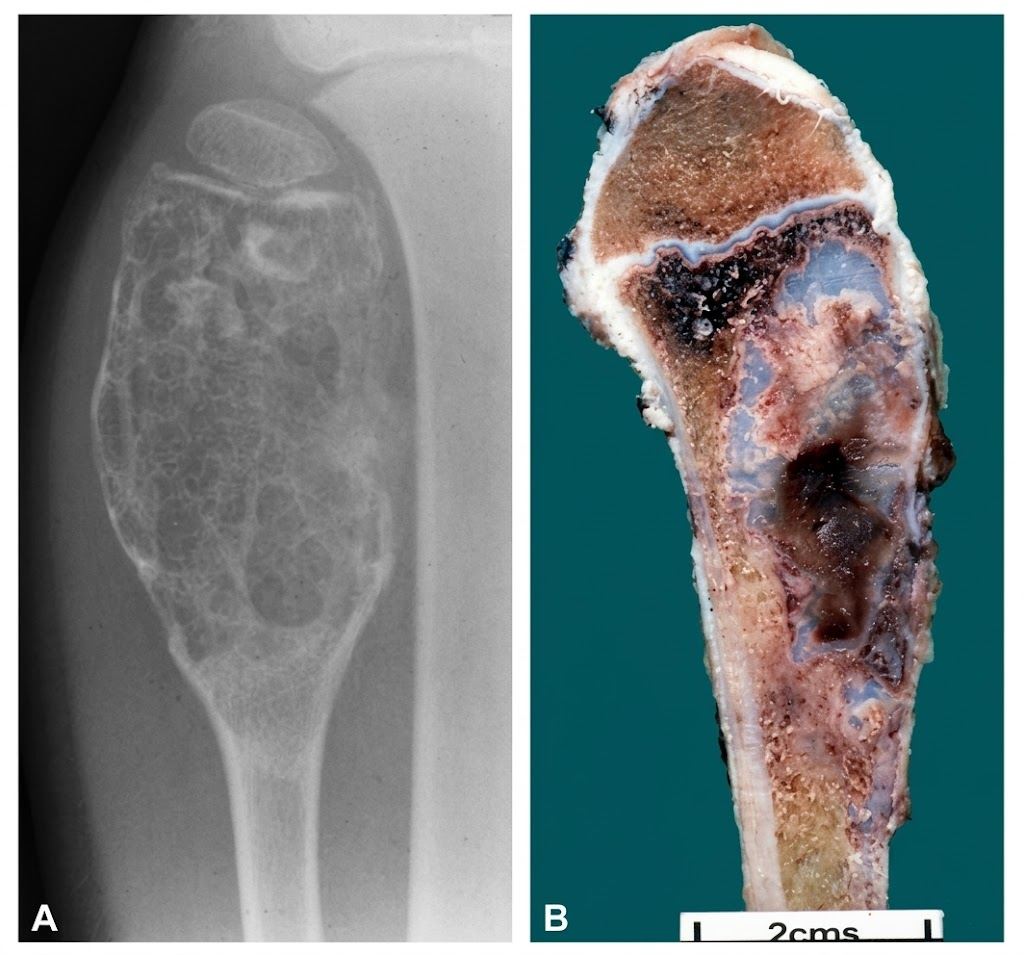
Hallazgo radiológico característico
0) En 1 minuto
Piensa en FCM cuando estás ante una lesión ósea lítica en un paciente joven, sobre todo en metáfisis de huesos largos, con un aspecto “fibroso” en la mayor parte de la lesión y pequeños focos cartilaginosos que, en histología, recuerdan a un cartílago de placa de crecimiento.
- ✅ Qué es: tumor óseo bifásico. Combina (1) nódulos de cartílago hialino benigno (con arquitectura “tipo placa de crecimiento”) y (2) un componente fusocelular fibroblástico (de aspecto “fibrosarcoma-like” pero, típicamente, de bajo grado).
- ✅ Por qué importa: es localmente agresivo. El riesgo clínico principal es la recidiva local si se trata con curetaje o resección incompleta.
- ✅ Claves diagnósticas: no suele haber atipia franca en el cartílago; y el rasgo más orientador del diagnóstico es la presencia de cartílago “epiphyseal plate-like” y osificación endocondral asociada.
- ✅ Tratamiento: la conducta más aceptada es la resección completa/en bloc cuando es técnicamente posible. Si se deja resección intralesional, el seguimiento debe ser estrecho y la reintervención se considera ante recidiva.
- ✅ Pronóstico: suele ser bueno si se consigue control local; en las series de referencia no se asocian metástasis ni muertes atribuibles al tumor, pero sí recidivas tras cirugía incompleta.
1) Conceptos generales
1.1 Terminología y clasificación OMS
El término OMS que conviene usar es fibrocartilaginous mesenchymoma. En español, “mesenquimoma fibrocartilaginoso” es una traducción aceptable. En la OMS 2020 se agrupa dentro de Other mesenchymal tumours of bone. Su codificación se ha aclarado en una corrección oficial del “blue book”, estableciendo ICD-O 8990/1 para esta entidad.
- Comportamiento: intermedio (localmente agresivo).
- Mensaje operativo: no se maneja como un sarcoma “de rutina”, pero tampoco como una lesión inocua tratable con legrado sin más; el punto crítico es el control local.
1.2 Historia y por qué se discute
Desde su descripción inicial, FCM ha generado debate porque comparte rasgos con entidades fibrosas benignas (y con su variante con cartílago, la llamada fibrocartilaginous dysplasia). En la práctica, lo más útil no es “ganar” la discusión nosológica, sino entender qué rasgos apuntan a FCM y qué consecuencias tiene: cuando no se extirpa completamente, recidiva.
2) Epidemiología
La evidencia epidemiológica procede de series pequeñas y casos aislados. Aun así, hay un patrón repetido: afecta sobre todo a niños, adolescentes y adultos jóvenes, con predilección por metáfisis de huesos largos.
- Edad: rango aproximado 9–25 años; en una serie de referencia (12 casos) y revisiones posteriores, la edad se concentra en la adolescencia.
- Sexo: discreto predominio masculino (no marcado).
- Localización: metáfisis de huesos largos; se ha descrito con frecuencia alrededor de la rodilla. En la serie clásica, la fíbula representa una proporción relevante de casos; también hay casos en pelvis y columna/rib (muy raros).
3) Clínica
La presentación suele ser poco espectacular. El síntoma típico es un dolor leve o intermitente de meses de evolución. A veces el paciente nota una tumefacción o una masa ósea, especialmente si hay expansión cortical o extensión yuxtacortical. No hay un síndrome constitucional característico.
- Dolor: mecánico o inespecífico, habitualmente de larga evolución.
- Masa / deformidad: ocasional, más visible en localizaciones superficiales.
- Limitación funcional: si está cerca de una articulación o en huesos de carga.
4) Imagen
4.1 Radiografía simple
Suele mostrar una lesión lítica, de patrón geográfico, en región metafisaria, a veces excéntrica. Puede haber adelgazamiento/expansión cortical y, en algunos casos, calcificaciones condroides. La reacción perióstica no suele ser prominente.
4.2 Tomografía computarizada (TC)
La TC es útil para caracterizar con precisión la cortical y para detectar mejor las calcificaciones de matriz condroide cuando existen. También ayuda a valorar una posible extensión yuxtacortical.
4.3 Resonancia magnética (RM)
La RM define la extensión intraósea y la relación con estructuras vecinas. Un dato orientador (no exclusivo) es observar un componente predominantemente fibroso con pequeños focos cartilaginosos. Es especialmente útil si hay sospecha de extensión a partes blandas o si se planifica resección en bloque.
5) Histopatología
En la práctica, el diagnóstico se apoya en un conjunto de rasgos relativamente constante: proliferación fusocelular blanda + nódulos de cartílago benigno + zonas con osificación endocondral “tipo placa de crecimiento”. Ese último punto (cartílago con arquitectura de placa de crecimiento) es el que más ayuda a separar FCM de otros tumores fibrosos.
5.1 Componente fusocelular
Predomina en muchas lesiones y puede ser el único componente muestreado en una biopsia pequeña. Suele ser relativamente hipocelular o moderadamente celular, con estroma colágeno y citología blanda. Aunque a veces se describe como “fibrosarcoma-like”, lo más importante es que no muestra el patrón de alto grado (mitosis atípicas, necrosis amplia, pleomorfismo marcado) que obligaría a replantear el diagnóstico.
5.2 Componente cartilaginoso
Aparece como nódulos de cartílago hialino sin atipia relevante. La característica más orientadora es que, en al menos parte del tumor, el cartílago puede organizarse en placas que recuerdan a la placa epifisaria, con osificación endocondral asociada.
6) Inmunohistoquímica
No existe un “marcador diagnóstico” específico de FCM. La inmunohistoquímica se utiliza sobre todo para apoyar la interpretación morfológica y para descartar miméticos cuando el muestreo es limitado.
-
Componente fusocelular:
- Vimentina: habitualmente positiva.
- SMA: puede ser focal/variable.
- Desmina: usualmente negativa (si es difusa, replantear diferencial miogénico).
- Ki-67: suele ser bajo (si es muy alto, revisar grado/diagnóstico).
-
Componente cartilaginoso:
- S100 / SOX9: positivos en condrocitos (como en cartílago benigno en general).
Nota práctica: cuando el diferencial incluye osteosarcoma de bajo grado, una prueba molecular para amplificación de MDM2 puede ser muy útil (más que una IHQ aislada).
7) Genética / pruebas dirigidas (cuándo ayudan)
La base molecular de FCM sigue sin estar definida por falta de series amplias. En la práctica, las pruebas moleculares se usan para excluir diagnósticos alternativos cuando la morfología o el contexto lo requieren:
- Si el diferencial es displasia fibrosa / fibrocartilaginous dysplasia: considerar GNAS (si la duda persiste).
- Si el diferencial es osteosarcoma de bajo grado: considerar MDM2 (amplificación) como apoyo.
- Si el diferencial es condrosarcoma mesenquimal: valorar pruebas de fusión específicas según protocolo del centro.
En resumen: en FCM la genética no suele “confirmar” el diagnóstico por sí sola; su utilidad es evitar errores en los casos con muestreo limitado o con rasgos atípicos.
8) Diagnóstico diferencial
8.1 Diferencial clínico-radiológico
| Entidad | Por qué se parece | Claves para separarla |
|---|---|---|
| Displasia fibrosa con cartílago (FCD) | Lesión fibro-ósea con nódulos condroides | Contexto y patrón típico de DF; genética (GNAS) puede ayudar si hay duda |
| Osteosarcoma de bajo grado (central/parostal) | Lesión fibrosa que puede ser engañosa | Buscar producción ósea tumoral y MDM2 (amplificación) cuando procede |
| Fibroma desmoplásico | Lesión fibrosa infiltrativa | No tiene el cartílago “tipo placa de crecimiento” |
| Condrosarcoma desdiferenciado | Componente condroide + sarcomatoso | El componente sarcomatoso es de alto grado; no hay cartílago con arquitectura de placa de crecimiento |
| Hamartoma condromesenquimal de pared torácica | Puede parecerse en costilla | Ocurre típicamente en lactantes; suele mostrar quistes tipo ABC y contexto distinto |
8.2 Diferencial histológico (lo que más confunde)
| Entidad | Trampa típica | Claves para separarla |
|---|---|---|
| FCM vs FCD | Ambas pueden tener componente fibroso y cartílago benigno | FCM: cartílago “tipo placa epifisaria” + conducta más recidivante cuando resección incompleta |
| FCM vs osteosarcoma de bajo grado | El fibroso “blando” puede confundirse | Buscar osteoide tumoral/producción ósea; apoyo con MDM2 si procede |
| FCM vs condrosarcoma desdiferenciado | Ambos son bifásicos | En desdiferenciado hay sarcoma de alto grado; FCM suele ser de bajo grado y cartílago tipo placa |
9) Tratamiento
9.1 Principio general: control local
La conducta se decide con un criterio simple: si el objetivo es minimizar recidiva, hay que evitar la resección incompleta. En la serie clásica y revisiones, la recidiva se asocia especialmente a curetaje / excisión intralesional. Por ello, cuando la localización lo permite, la opción más sólida es resección completa/en bloc.
9.2 Cirugía y reconstrucción
- Resección completa/en bloc: preferible cuando es factible, especialmente en lesiones recidivadas o con rotura cortical/masa yuxtacortical.
- Curetaje: se considera de mayor riesgo de recidiva; si se realiza, el seguimiento debe ser estrecho y el umbral de reintervención bajo.
- Reconstrucción: dependerá del hueso afectado y del defecto (injerto, prótesis, osteosíntesis, etc.).
9.3 Radioterapia y quimioterapia
No existe un papel estándar para radioterapia o quimioterapia en FCM. Si un caso se plantea como candidato a terapia sistémica, lo correcto es reconfirmar el diagnóstico (morfología + revisión experta + pruebas dirigidas) porque, en general, el problema de FCM es el control local.
10) Pronóstico y seguimiento
El pronóstico global suele ser bueno cuando se consigue resección completa. Lo que condiciona el curso es la recidiva local. En la literatura de referencia se insiste en que la recidiva aparece sobre todo tras cirugía incompleta, y que no hay metástasis ni muertes atribuibles al tumor en las series más citadas.
- Riesgo principal: recidiva local tras curetaje/excisión intralesional.
- Metástasis: no es un rasgo típico (no documentada en series de referencia).
- Seguimiento (práctico):
- 0–2/3 años: control clínico + imagen local cada 4–6 meses (según tipo de resección y localización).
- Hasta 5 años: anual si estabilidad.
- Torácico: no rutinario si no hay motivos clínicos; individualizar según comité.
11) Algoritmo diagnóstico y terapéutico
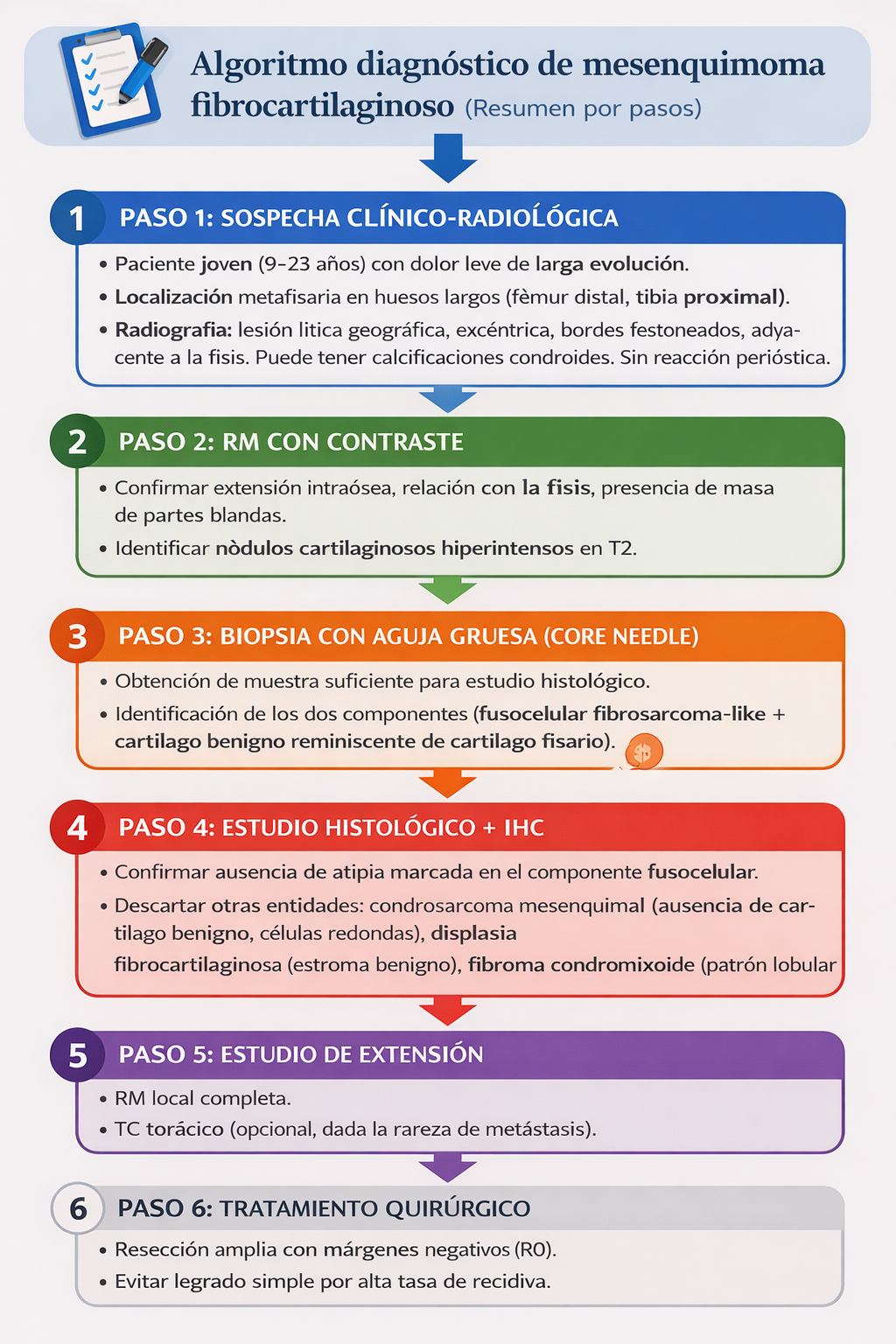
📌 Resumen para la práctica clínica
Indicaciones
- ✅ Lesión lítica metafisaria en paciente joven con componente fibroso predominante y focos cartilaginosos.
- ✅ Confirmación histológica de patrón bifásico (fusocelular + cartílago tipo placa de crecimiento).
- ✅ Necesidad de control local por riesgo de recidiva tras resección incompleta.
Técnica
- 🔧 Imagen: RX + TC (cortical/calcificaciones) + RM (extensión y planificación).
- 🔧 Biopsia dirigida para muestrear componente cartilaginoso, evitando falsos diagnósticos por muestreo parcial.
- 🔧 Cirugía: resección completa/en bloc cuando es factible; considerar reconstrucción según defecto.
Riesgos
- ⚠️ Biopsia no representativa (solo componente fibroso) con diagnóstico erróneo.
- ⚠️ Tratamiento intralesional/curetaje con riesgo aumentado de recidiva local.
- ⚠️ Confusión con osteosarcoma de bajo grado o con fibrocartilaginous dysplasia.
Resultados
- ✅ Pronóstico global favorable si se logra control local.
- ✅ Recidiva local principalmente asociada a resección incompleta; metástasis no típica en series de referencia.
12) Bibliografía
📘 OMS / clasificación
- 📄 WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. 5th ed. Lyon: IARC; 2020.
- 📄 IARC/WHO. Soft Tissue and Bone Tumours — Corrections. (Corrección ICD-O: 8990/1 “Fibrocartilaginous mesenchymoma”).
📑 Trabajos clave
- 📄 Dahlin DC, Bertoni F, Beabout JW, Campanacci M. Fibrocartilaginous mesenchymoma with low-grade malignancy. Skeletal Radiol. 1984;12:263–269.
- 📄 Bulychova IV, Unni KK, Bertoni F, Beabout JW. Fibrocartilagenous mesenchymoma of bone. Am J Surg Pathol. 1993;17:830–836. (Recidiva tras excisión intralesional; sin metástasis ni muertes atribuibles).
- 📄 Oh SJ, et al. Fibrocartilaginous mesenchymoma with an unusual location in the rib. J Pathol Transl Med. 2020. (Revisión y claves diagnósticas; “<35 casos”).
- 📄 Al-Khudairi R, et al. Fibrocartilaginous mesenchymoma of the rib with atypical imaging features. BJR Case Rep. 2025. (Pitfall de biopsia; miméticos y radiología).
- 📄 Cherradi N, et al. Fibrocartilaginous mesenchymoma of bone. A case report. Rev Chir Orthop Reparatrice Appar Mot. 1999. (Discusión del comportamiento y diferencial; “metástasis no reportadas”).