Leiomiosarcoma óseo primario — Actualización 2026: sarcoma de músculo liso de estirpe vascular, diagnóstico de exclusión y manejo multidisciplinar
El leiomiosarcoma (LMS) primario del hueso es una neoplasia maligna extremadamente infrecuente, que representa menos del 0,2% de todos los tumores óseos primarios. Deriva de las células musculares lisas de la pared vascular intraósea o, alternativamente, de células madre mesenquimales con capacidad de diferenciación miogénica. Durante décadas confundido con metástasis de leiomiosarcomas uterinos, retroperitoneales o de partes blandas, su diagnóstico exige hoy una rigurosa exclusión clínica, radiológica e inmunohistoquímica de un tumor primario extraesquelético. La clasificación de la Organización Mundial de la Salud (OMS 2020) lo integra dentro del grupo de «tumores de estirpe incierta» con diferenciación muscular lisa, y establece como criterio diagnóstico la positividad para actina de músculo liso (SMA), caldesmón y desmina (esta última en el 50-60% de los casos), con negatividad para marcadores epiteliales (citoqueratinas, EMA) y S100. La Actualización 2026 incorpora los avances en genética molecular (pérdida de RB1, mutaciones en TP53, alteraciones en MYOCD), la reclasificación de los antiguos «hemangiopericitomas» como tumores fibrosos solitarios (STAT6+) y la creciente evidencia sobre la eficacia de los inhibidores de tirosina cinasa en enfermedad avanzada. Esta ficha desarrolla de manera extensa y fundamentada la epidemiología, clínica, radiología, histopatología, diagnóstico diferencial y opciones terapéuticas actuales de esta entidad, con especial énfasis en los criterios de exclusión que permiten diferenciarla del leiomiosarcoma metastásico.
En 1 minuto
Sarcoma óseo primario de alto grado, derivado de células musculares lisas. <0,2% de tumores óseos primarios.
Diagnóstico de exclusión. Descartar LMS metastásico de útero, retroperitoneo, tubo digestivo o partes blandas.
Dolor progresivo, masa palpable. Edad 40-60 años, predominio masculino (2:1). Localización: metáfisis de fémur distal, tibia proximal.
Lesión lítica, geográfica o permeativa, sin matriz mineralizada. Reacción perióstica variable. TC torácico obligatorio.
Positivo: SMA (>95%), caldesmón (80-90%), desmina (50-60%). Negativo: S100, STAT6, CD34, citoqueratinas.
Pérdida de RB1 (70%), mutaciones TP53 (40%), amplificación MYOCD (30%). Sin translocación específica.
Resección quirúrgica amplia (R0). RT adyuvante en alto riesgo. QT con antraciclinas + ifosfamida en metastásico.
SG a 5 años: 50-60%. Factores adversos: >50 años, >8 cm, grado III, márgenes positivos, metástasis al diagnóstico.
Conceptos generales
1.1 Definición y nosología
El leiomiosarcoma (LMS) es un sarcoma maligno compuesto por células con diferenciación de músculo liso. Cuando se origina en el hueso sin evidencia de tumor primario extraesquelético, se denomina leiomiosarcoma óseo primario. Es una entidad excepcional, mucho menos frecuente que el LMS de partes blandas (3-5% de todos los sarcomas de partes blandas) o el LMS uterino.
La clasificación de la Organización Mundial de la Salud (OMS 2020) incluye el LMS dentro del grupo de «tumores de estirpe incierta» con diferenciación muscular lisa. No existe una categoría separada para el LMS óseo; se clasifica dentro de los sarcomas óseos productores de colágeno, junto con el fibrosarcoma y el sarcoma pleomórfico indiferenciado (UPS).
Durante décadas, muchos casos publicados como LMS óseo primario fueron, en realidad, metástasis de LMS uterinos, retroperitoneales o de partes blandas. El advenimiento de la inmunohistoquímica y, más recientemente, de la genética molecular, ha permitido establecer criterios diagnósticos más estrictos que han reducido drásticamente su incidencia real.
1.2 Histogénesis
El origen del LMS óseo primario es controvertido. Se han propuesto dos teorías principales:
- Origen vascular: La rica vascularización intraósea sugiere que el tumor podría originarse en las células musculares lisas de las paredes de los vasos sanguíneos. El frecuente hallazgo de un patrón hemangiopericitomatoso en algunas áreas apoyaría esta hipótesis.
- Origen en célula madre mesenquimal: Las células madre multipotentes de la médula ósea podrían sufrir una transformación neoplásica con diferenciación miogénica. Explicaría la ausencia de un precursor vascular evidente en algunos casos.
En la actualidad, se acepta que ambas vías son posibles. El LMS óseo, el LMS de partes blandas y el LMS uterino comparten alteraciones genéticas similares (pérdida de RB1, mutaciones TP53), lo que sugiere un mecanismo patogénico común independiente del tejido de origen.
Epidemiología
2.1 Incidencia
- Frecuencia: El LMS representa menos del 0,2% de todos los tumores óseos primarios. En las series más amplias del Rizzoli (2019) y del Mayo Clinic (2022), apenas se identificaron 30-40 casos en 30-40 años de seguimiento.
- Proporción respecto al LMS global: El LMS óseo primario constituye menos del 1% de todos los LMS, frente al LMS de partes blandas (70%), uterino (15-20%) y cutáneo (5-10%).
2.2 Edad y sexo
- Edad: Rango amplio (9-80 años), pero muy raro antes de los 20 años. Pico de incidencia en la 5ª-6ª décadas (mediana 45-50 años).
- Sexo: Predominio masculino en todas las series, con una relación hombre:mujer de 2:1. Este predominio es inverso al LMS uterino (exclusivamente femenino) y contrasta con el LMS de partes blandas (ligero predominio femenino).
2.3 Localización
- Huesos largos (80%):
- Fémur distal (30-40%)
- Tibia proximal (15-20%)
- Húmero proximal (10-15%)
- Fémur proximal, peroné, radio, cúbito (casos aislados)
- Localizaciones axiales (20%): Pelvis (ilíaco, sacro), columna vertebral, costillas, clavícula, mandíbula, esternón.
- Localización dentro del hueso: Predominantemente metafisaria, con extensión frecuente a la epífisis y diáfisis en estadios avanzados.
Clínica
- Dolor: Síntoma de presentación en >90% de los pacientes. Insidioso al inicio, progresivo, a menudo nocturno. Duración media: 4-8 meses antes del diagnóstico.
- Tumoración palpable: Presente en el 40-60% de los casos, especialmente cuando el tumor ha roto la cortical y se extiende a partes blandas.
- Fractura patológica: Entre el 10% y el 20% debutan como fractura. La incidencia es menor que en osteosarcoma o condrosarcoma debido al patrón lítico expansivo.
- Limitación funcional: Cojera, impotencia funcional, derrame articular (localizaciones yuxtaarticulares).
- Síntomas B: Fiebre, pérdida de peso, astenia (infrecuentes, sugieren enfermedad metastásica).
Imagen
4.1 Radiografía simple
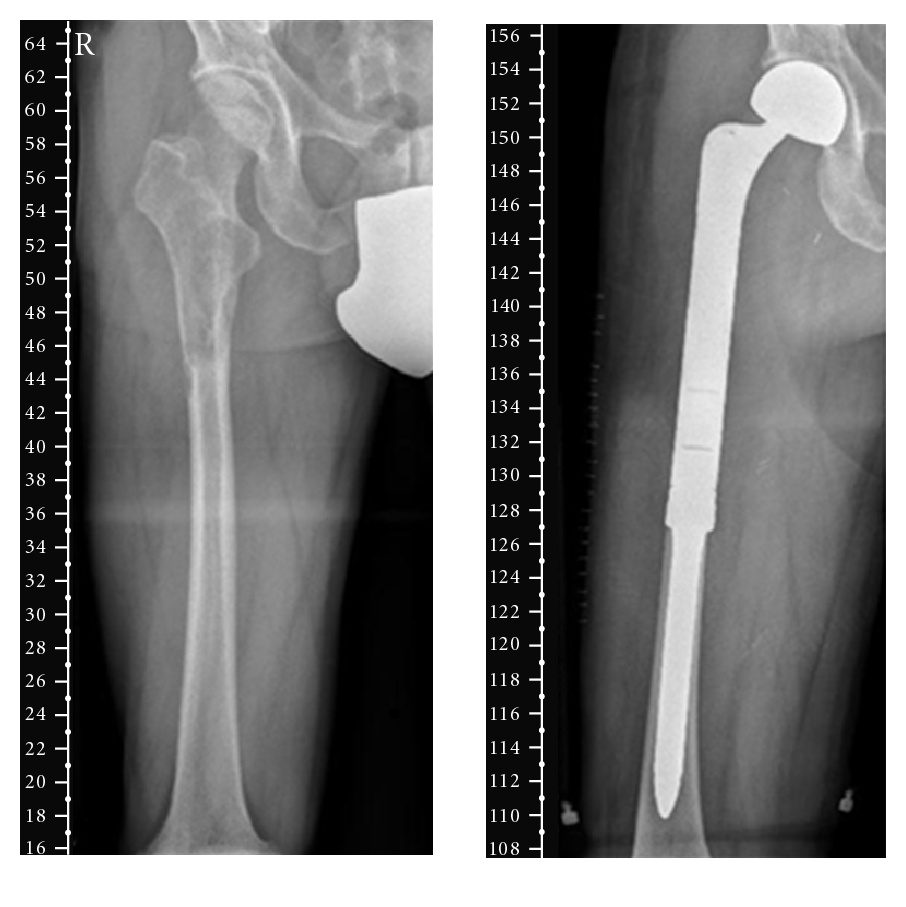
- Patrón de destrucción ósea: Variable y en relación con el grado histológico. En general, cuanto más agresivo es el tumor, más amplia es la zona de transición y más marcada la destrucción cortical.
- LMS de bajo grado: Lesión lítica geográfica, con bordes mal definidos o parcialmente escleróticos. Puede simular un tumor de células gigantes, una metástasis lítica o un linfoma óseo.
- LMS de alto grado: Patrón permeativo o moteado, con zona de transición ancha, destrucción cortical y frecuente extensión a partes blandas.
- Matriz tumoral: No hay mineralización de la matriz, lo que ayuda a diferenciarlo de osteosarcoma y condrosarcoma. Pueden verse calcificaciones distróficas en áreas de necrosis, pero no constituyen matriz tumoral verdadera.
- Reacción perióstica: Presente en aproximadamente el 50-60% de los casos. Puede ser sólida en lesiones de crecimiento más lento o interrumpida (Codman, espículas) en tumores de alto grado. No es específica, pero refuerza la impresión de agresividad.
- Cortical y partes blandas: La rotura cortical y la masa de partes blandas son hallazgos frecuentes, sobre todo en tumores de alto grado. La radiografía puede sugerirla, pero su extensión se valora mejor en RM.
- Diagnóstico diferencial: Radiográficamente puede solaparse con metástasis líticas, linfoma óseo, sarcoma pleomórfico indiferenciado del hueso y, en algunos casos, tumor de células gigantes. La radiografía orienta, pero no permite un diagnóstico específico sin correlación histológica.
4.2 Resonancia magnética (RM)
- T1: Señal isointensa o ligeramente hipointensa respecto al músculo.
- T2/STIR: Señal heterogénea, predominantemente hiperintensa. Las áreas de necrosis y hemorragia son muy hiperintensas; las zonas de mayor celularidad pueden ser hipointensas.
- T1 con contraste: Realce intenso y heterogéneo. El patrón de realce puede ser periférico (en anillo) en tumores con necrosis central.
- Difusión (DWI): Restricción en áreas celulares (ADC bajo), útil para guiar la biopsia.
- Extensión: La RM es esencial para evaluar la extensión intraósea, la afectación de partes blandas y la relación con estructuras neurovasculares. El LMS suele mostrar un patrón infiltrativo, sin seudocápsula bien definida.
4.3 Tomografía computarizada (TC)
- Estadificación local: Útil en localizaciones complejas (pelvis, columna, sacro).
- Matriz tumoral: Confirma la ausencia de mineralización de matriz.
- TC torácico: OBLIGATORIO. El pulmón es el sitio predominante de metástasis (70-80%). Hasta el 15-20% de los pacientes tienen metástasis sincrónicas al diagnóstico.
4.4 PET-TC con FDG
- Captación: El LMS suele ser FDG-ávido, con SUVmax elevado, en general mayor en tumores de alto grado.
- Correlación: El SUVmax tiende a relacionarse con grado histológico, celularidad, necrosis e índice proliferativo (Ki67).
- Utilidad: Útil para estadificación, detección de metástasis a distancia y valoración de respuesta metabólica al tratamiento.
- Limitaciones: No es específica; debe interpretarse junto con RM, TC y biopsia.
Histopatología
5.1 Microscopía óptica
- Arquitectura: Fascículos entrelazados de células fusiformes (patrón «en espiga de trigo»). Áreas bien diferenciadas: células con citoplasma eosinófilo y núcleos en «cigarro puro». Áreas poco diferenciadas: células redondas, epitelioides o pleomórficas. En 20-30% de los casos se observan áreas con patrón hemangiopericitomatoso (no confundir con SFT).
- Citología: Células fusiformes con citoplasma eosinófilo, fibrilar. Núcleos alargados, hipercromáticos, con extremos romos (característico). Pleomorfismo variable.
- Estroma: Escaso en áreas hipercelulares; colágeno hialino en áreas hipocelulares (puede simular osteoide).
- Mitosis y necrosis: Actividad mitótica elevada en LMS de alto grado (>10 mitosis/10 HPF). Necrosis geográfica frecuente (50-60%).
5.2 Gradación histológica (FNCLCC)
El LMS óseo se gradúa según el sistema de la Federación Francesa de Centros de Lucha contra el Cáncer (FNCLCC):
| Parámetro | Puntuación |
|---|---|
| Diferenciación tumoral | Bien diferenciado: 2 puntos; poco diferenciado: 3 puntos |
| Índice mitótico | 0-9/10 HPF: 1; 10-19: 2; ≥20: 3 |
| Necrosis tumoral | 0%: 0; <50%: 1; ≥50%: 2 |
- Grado 1 (bajo): 2-3 puntos
- Grado 2 (intermedio): 4-5 puntos
- Grado 3 (alto): 6-8 puntos
A diferencia del LMS de partes blandas, el grado histológico no ha demostrado correlación estadísticamente significativa con la supervivencia en el LMS óseo, probablemente debido al reducido número de casos.
5.3 Microscopía electrónica
- Hallazgos ultraestructurales: Filamentos de actina con cuerpos densos (diagnóstico), lámina basal externa, vesículas de pinocitosis, uniones intercelulares de tipo adherente.
- Utilidad actual: La microscopía electrónica ha sido desplazada por la inmunohistoquímica. Actualmente solo se emplea en casos excepcionales con IHC equívoca.
Inmunohistoquímica (IHC)
La IHC es imprescindible para el diagnóstico y el diagnóstico diferencial. Sin un panel completo, no es posible confirmar el LMS ni excluir otras neoplasias fusocelulares.
6.1 Marcadores positivos
- Actina de músculo liso (SMA): Positiva en >95% de los casos. Patrón citoplasmático difuso. Es el marcador más sensible.
- Caldesmón (h-caldesmón): Positivo en 80-90% de los casos. Marcador específico de diferenciación muscular lisa (no se expresa en miofibroblastos). Muy útil para diferenciar LMS de sarcomas miofibroblásticos.
- Desmina: Positiva en 50-60% de los casos. Menos sensible que SMA, pero más específica. El patrón puede ser citoplasmático difuso o en cuerpos densos.
- Vimentina: Positiva en 100% de los casos. Inespecífica.
- Actina músculo específica (HHF35): Positiva en 70-80% de los casos. Actualmente poco utilizada.
- CKIT (CD117): Positivo en 10-15% de los casos (focal, débil). No debe confundirse con GIST (que es DOG1+, con mutación KIT/PDGFRA).
- EMA: Positivo focal en <10% de los casos (formas epitelioides).
6.2 Marcadores negativos
- S100 y SOX10: Negativos (diferencia de tumor de vaina nerviosa, melanoma).
- STAT6: Negativo (diferencia de tumor fibroso solitario).
- CD34: Negativo (puede ser positivo en áreas hemangiopericitomatosas, pero no en las células fusiformes).
- Citoqueratinas (AE1/AE3, CAM5.2): Negativas (diferencia de carcinoma sarcomatoide, sarcoma sinovial).
- Miogenina y MyoD1: Negativos (diferencia de rabdomiosarcoma).
- MDM2 y CDK4: Negativos (diferencia de liposarcoma desdiferenciado).
- ERG, CD31, FLI1: Negativos (diferencia de angiosarcoma).
6.3 Marcadores de proliferación y pronóstico
- Ki67 (MIB-1): Variable (10-50% en LMS de alto grado). Se correlaciona con el grado histológico y el pronóstico.
- p53: Sobreexpresión nuclear en el 40-50% de los casos (asociada a mutación TP53). Se correlaciona con peor pronóstico.
- RB1: Pérdida de expresión nuclear en el 70% de los casos (deleción/loss of function).
6.4 Panel IHC recomendado
Panel mínimo para diagnóstico y diferencial: SMA, caldesmón, desmina, S100, STAT6, CD34, citoqueratinas, EMA, miogenina, MDM2.
Genética molecular
7.1 Alteraciones cromosómicas
- Cariotipo complejo: El LMS óseo, al igual que el LMS de partes blandas, presenta cariotipos complejos con numerosas ganancias y pérdidas cromosómicas, sin translocaciones balanceadas recurrentes.
- Pérdida de 10q, 13q, 17p: Frecuentes, corresponden a la pérdida de genes supresores tumorales.
7.2 Genes implicados
- RB1 (13q14): Pérdida de función (deleción, mutación, hipermetilación) en 70-80% de los LMS. Es la alteración genética más frecuente. La pérdida de expresión nuclear de RB1 por IHC es un marcador útil.
- TP53 (17p13): Mutación o deleción en 40-50% de los casos. Se correlaciona con alto grado y mal pronóstico.
- MYOCD (17p12): Ganancia/amplificación en 30% de los LMS. MYOCD codifica un coactivador transcripcional de la diferenciación muscular lisa. La amplificación de MYOCD es un evento oncogénico temprano en LMS.
- ATRX (Xq21): Mutaciones en 20-30% de los LMS (especialmente en formas pediátricas).
- PTEN (10q23): Pérdida o mutación en 15-20%.
7.3 Utilidad clínica
- No hay alteración genética específica para el diagnóstico. No es posible confirmar LMS por genética.
- La detección de pérdida de RB1 puede ser útil en el diagnóstico diferencial con sarcomas que conservan RB1.
- La presencia de amplificación de MYOCD es un marcador de diferenciación muscular lisa, pero no es 100% específico.
- Las alteraciones de TP53 y RB1 tienen valor pronóstico.
- Biopsia líquida: En investigación. La detección de ctDNA con alteraciones en TP53 y RB1 podría ser útil para monitorizar la respuesta al tratamiento y detectar recidivas precoces.
Diagnóstico diferencial
El diagnóstico diferencial del LMS óseo primario es muy amplio e incluye tanto neoplasias primarias como metastásicas. La correcta clasificación requiere la integración de la clínica, la radiología, la IHC y la genética molecular.
8.1 Diferencial radiológico
| Entidad | Claves radiológicas |
|---|---|
| Fibrosarcoma óseo | Idéntico. Solo diferenciación IHC. |
| Sarcoma pleomórfico indiferenciado (UPS) | Idéntico. Solo diferenciación IHC. |
| Linfoma óseo | Patrón permeativo, masa de partes blandas extensa, edad >50 años. |
| Metástasis lítica | Múltiples lesiones, edad >50 años, antecedente oncológico. |
| Sarcoma de Ewing | Diáfisis, paciente joven, reacción perióstica en capas de cebolla. |
| Tumor de células gigantes | Epífisis, paciente 20-40 años, márgenes geográficos. |
8.2 Diferencial histológico e IHC
| Entidad | IHC positiva | IHC negativa | Genética |
|---|---|---|---|
| Leiomiosarcoma metastásico | SMA, caldesmón, desmina (50%) | Idéntico al primario | Idéntico |
| Fibrosarcoma | Vimentina | SMA, caldesmón, desmina | Complejo |
| Sarcoma pleomórfico indiferenciado | Vimentina | SMA, caldesmón, desmina, S100, EMA | Complejo |
| Tumor fibroso solitario (SFT) | STAT6, CD34 | SMA, desmina, S100 | NAB2-STAT6 |
| Sarcoma sinovial | Citoqueratinas, EMA, TLE1, CD99 | SMA, desmina, S100 | SS18-SSX1/2 |
| Rabdomiosarcoma | Miogenina, MyoD1, desmina | SMA, caldesmón | PAX3/7-FOXO1 (alveolar) |
| Liposarcoma desdiferenciado | MDM2, CDK4 | SMA, desmina, S100 | Amplificación MDM2 |
| Melanoma amelánico | S100, SOX10, HMB45, MelanA | SMA, desmina | BRAF, NRAS, KIT |
| Carcinoma sarcomatoide | Citoqueratinas, EMA | SMA, desmina | Complejo |
Criterios de exclusión: leiomiosarcoma óseo primario vs. metastásico
EL LMS ÓSEO PRIMARIO ES UN DIAGNÓSTICO DE EXCLUSIÓN
9.1 Criterios mayores (obligatorios)
- Exclusión clínica de tumor primario extraesquelético:
- Anamnesis exhaustiva: histerectomía previa (aunque fuera por miomas), cirugías de partes blandas, antecedente de tumores retroperitoneales o gastrointestinales.
- Exploración física: examen pélvico (mujeres), tacto rectal (varones), palpación abdominal.
- Pruebas de imagen: TC abdominopélvico, RM pélvica, ecografía transvaginal si es necesario.
- Revisión de piezas quirúrgicas previas:
- Toda paciente con LMS óseo y antecedente de histerectomía debe tener las piezas de histerectomía revisadas por un patólogo experimentado. Los miomas catalogados como benignos pueden albergar focos de LMS de bajo grado no diagnosticados.
- Predominio óseo:
- El tumor debe afectar predominantemente al hueso, con nula o mínima afectación de partes blandas (<30% del volumen total).
- Un LMS que surge en partes blandas y secundariamente afecta al hueso adyacente (>70% del tumor en partes blandas) debe clasificarse como LMS de partes blandas con invasión ósea, no como LMS óseo primario.
9.2 Criterios menores (orientadores)
- Edad y sexo: Varón >40 años (el LMS uterino es casi exclusivo de mujeres). Un LMS óseo en una mujer joven debe hacer sospechar fuertemente metástasis de LMS uterino.
- Localización única: El LMS óseo primario suele ser solitario; las lesiones múltiples son mucho más sugestivas de metástasis.
- Tamaño: Los LMS metastásicos suelen ser más pequeños y múltiples.
Tratamiento
El tratamiento del LMS óseo primario sigue los mismos principios que el de otros sarcomas óseos de alto grado, aunque la evidencia se limita a series de casos retrospectivas.
10.1 Cirugía
- Resección amplia (R0): Es el estándar y el único tratamiento potencialmente curativo. Debe realizarse con márgenes de resección de 2-3 cm en hueso y 1-2 cm en partes blandas, o fascia intacta como barrera.
- Cirugía preservadora de extremidad: Posible en >90% de los casos. La planificación debe basarse en la RM preoperatoria e incluir la resección del trayecto de biopsia.
- Amputación: Reservada para casos con infiltración neurovascular masiva no reconstructible, múltiples recidivas o fracaso de cirugías previas.
10.2 Radioterapia
- Adyuvante: Indicada en tumores de alto grado (grado 3), márgenes positivos (R1/R2), localización anatómica que impide resección amplia (pelvis, columna, base de cráneo) y recidivas. Dosis: 60-66 Gy en fraccionamiento convencional.
- Neoadyuvante: Puede considerarse en tumores grandes (>10 cm) o borderline resecables para reducir el volumen tumoral y facilitar la cirugía. Dosis: 50 Gy.
- Protonterapia/Adroterapia: En localizaciones complejas (columna, pelvis, base de cráneo). Ofrece mejor control local con menor toxicidad.
10.3 Quimioterapia
- Neoadyuvante/adyuvante: No hay consenso. No se ha demostrado beneficio en supervivencia global en ensayos aleatorizados. Puede considerarse en tumores de alto grado, >8 cm o con quimiosensibilidad conocida. El régimen más utilizado es doxorrubicina 75 mg/m² + ifosfamida 9-10 g/m².
- Enfermedad metastásica:
- Primera línea: Antraciclinas (doxorrubicina sola o combinada). Tasas de respuesta: 15-25%.
- Segunda línea: Pazopanib (aprobado en sarcomas de partes blandas). Evidencia limitada en LMS óseo, pero extrapolable del LMS de partes blandas. Tasas de respuesta: 5-10%, beneficio en SLE. Gemcitabina + docetaxel (respuestas del 15-20%). Trabectedina (actividad modesta).
- Inmunoterapia: Pembrolizumab y nivolumab muestran tasas de respuesta bajas (<10%) en LMS. No aprobados como estándar.
Pronóstico y seguimiento
11.1 Pronóstico
- Supervivencia global a 5 años: 50-60% (series de centros de referencia).
- Supervivencia global a 10 años: 30-40%.
- Factores pronósticos adversos:
- Edad >50 años.
- Tamaño >8 cm.
- Grado histológico III (FNCLCC).
- Márgenes quirúrgicos positivos (R1/R2).
- Metástasis al diagnóstico (15-20%).
- Localización axial (pelvis, columna).
- Fractura patológica (controvertido).
- Recidiva local: 20-30% a los 5 años. La mayoría ocurren en los primeros 2-3 años.
- Metástasis a distancia: Principal causa de muerte. 40-50% de los pacientes desarrollarán metástasis, con una mediana de aparición de 12-24 meses. Localización predominante: pulmón (80%), hueso (15%), hígado (10%).
11.2 Seguimiento sugerido
- Primeros 2-3 años: RM/TC de la región primaria cada 3-4 meses. TC torácico cada 3-6 meses.
- Años 2-5: RM/TC cada 6 meses. TC torácico cada 6 meses.
- Años 5-10: RM/TC anual. TC torácico anual.
- Más de 10 años: Individualizar. El riesgo de recidiva tardía justifica seguimiento prolongado en pacientes de alto riesgo.
Algoritmo diagnóstico
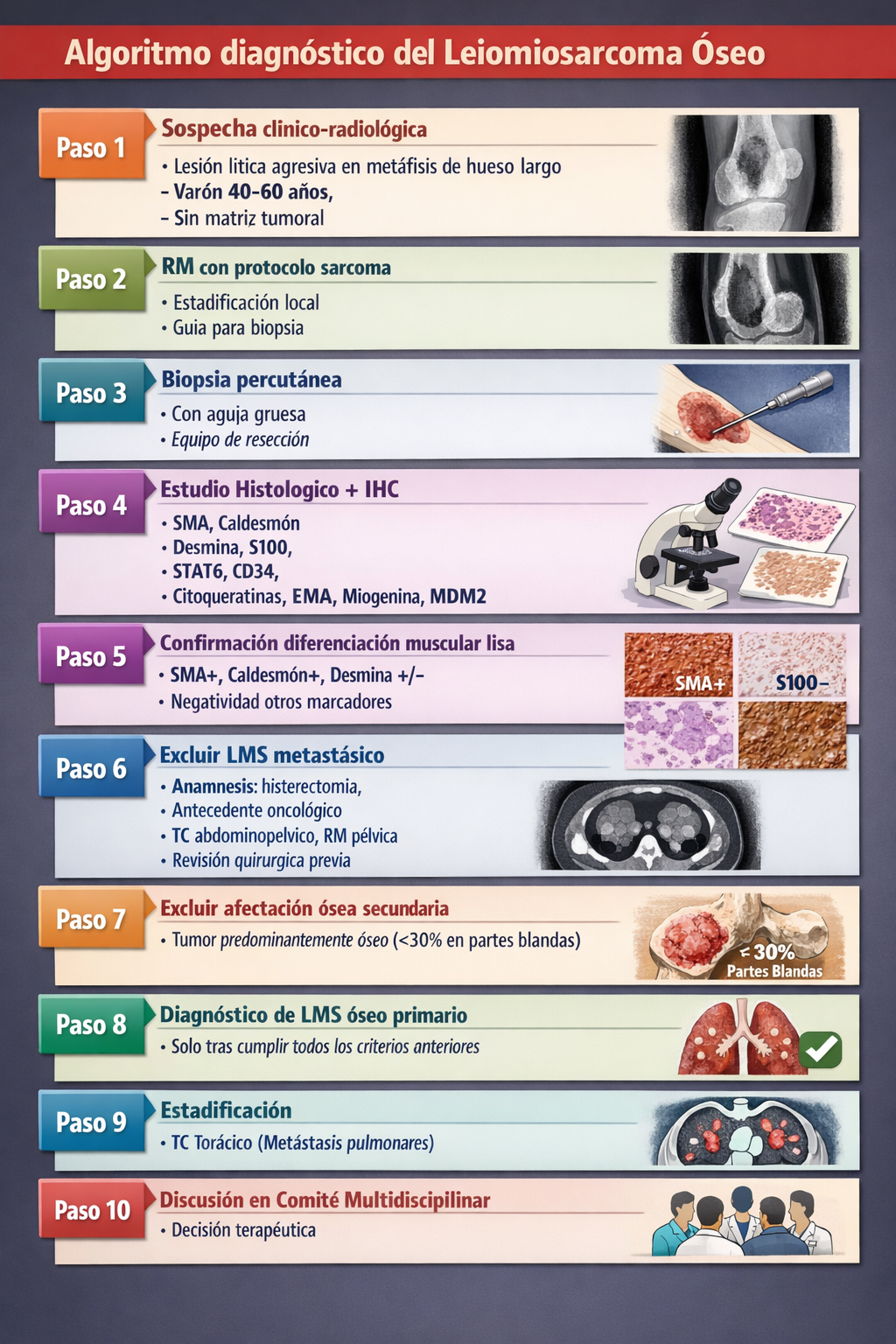
Resumen práctico
Indicaciones
- Resección quirúrgica amplia con márgenes negativos (R0) en todo LMS óseo localizado resecable.
- Radioterapia adyuvante en tumores de alto grado, márgenes positivos o localización anatómica desfavorable.
- Quimioterapia con antraciclinas ± ifosfamida en primera línea de enfermedad metastásica.
- Pazopanib en segunda línea de enfermedad metastásica (fracaso a antraciclinas).
Técnica
- Cirugía: resección En bloc con margen de 2-3 cm en hueso y 1-2 cm en partes blandas. Trayecto de biopsia excindible.
- Radioterapia: IMRT, protonterapia o adroterapia según localización. Dosis: 60-66 Gy adyuvante, 50 Gy neoadyuvante.
- Quimioterapia: doxorrubicina 75 mg/m² + ifosfamida 9 g/m² cada 21 días, 6 ciclos.
- Pazopanib: 800 mg/día vía oral.
Riesgos y complicaciones
- Diagnosticar como primario un LMS metastásico no detectado (error más grave).
- Resección marginal (excisión casual) de LMS no diagnosticado.
- Márgenes positivos (R1/R2) que aumentan el riesgo de recidiva local.
- Metástasis pulmonares (30-40% de los pacientes).
- Mala respuesta a quimioterapia en enfermedad avanzada.
Qué esperar del resultado
- Supervivencia global a 5 años: 50-60%.
- Supervivencia libre de recidiva local a 5 años: 70-80% (cirugía R0 + RT).
- Supervivencia libre de metástasis a 5 años: 50-60%.
Resumen para la práctica clínica
📌 Indicaciones
- ✓ Resección quirúrgica amplia con márgenes negativos (R0) en todo LMS óseo localizado resecable.
- ✓ Radioterapia adyuvante en tumores de alto grado, márgenes positivos o localización anatómica desfavorable.
- ✓ Quimioterapia con antraciclinas ± ifosfamida en primera línea de enfermedad metastásica.
- ✓ Pazopanib en segunda línea de enfermedad metastásica (fracaso a antraciclinas).
🔧 Técnica
- 🔧 Cirugía: resección En bloc con margen de 2-3 cm en hueso y 1-2 cm en partes blandas. Trayecto de biopsia excindible.
- 🔧 Radioterapia: IMRT, protonterapia o adroterapia según localización. Dosis: 60-66 Gy adyuvante, 50 Gy neoadyuvante.
- 🔧 Quimioterapia: doxorrubicina 75 mg/m² + ifosfamida 9 g/m² cada 21 días, 6 ciclos.
- 🔧 Pazopanib: 800 mg/día vía oral.
⚠️ Riesgos
- ⚠️ Diagnosticar como primario un LMS metastásico no detectado (error más grave).
- ⚠️ Resección marginal (excisión casual) de LMS no diagnosticado.
- ⚠️ Márgenes positivos (R1/R2) que aumentan el riesgo de recidiva local.
- ⚠️ Metástasis pulmonares (30-40% de los pacientes).
- ⚠️ Mala respuesta a quimioterapia en enfermedad avanzada.
✅ Resultados
- ✅ Supervivencia global a 5 años: 50-60%.
- ✅ Supervivencia libre de recidiva local a 5 años: 70-80% (cirugía R0 + RT).
- ✅ Supervivencia libre de metástasis a 5 años: 50-60%.
Bibliografía
Clasificación y guías clínicas
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon: IARC Press; 2020. (Capítulo: Leiomyosarcoma of bone, p. 456-458).
- Strauss SJ, Frezza AM, Abecassis N, et al. Bone sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2024;35(8):678-695. doi:10.1016/j.annonc.2024.03.008
- von Mehren M, Kane JM, Agulnik M, et al. Soft Tissue Sarcoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024;22(4):249-266.
Series clínicas y epidemiología
- Brewer P, Sumathi V, Grimer RJ, et al. Primary leiomyosarcoma of bone: analysis of 26 cases from a single institution. Bone Joint J. 2019;101-B(9):1129-1134. doi:10.1302/0301-620X.101B9.BJJ-2019-0123.R1
- Recine F, Bongiovanni A, Casadei R, et al. Primary leiomyosarcoma of the bone: a retrospective study from the Rizzoli experience. Clin Sarcoma Res. 2020;10:12. doi:10.1186/s13569-020-00134-w
- Antonescu CR, Erlandson RA, Huvos AG. Primary leiomyosarcoma of bone: a clinicopathologic, immunohistochemical, and ultrastructural study of 33 cases and a literature review. Am J Surg Pathol. 1997;21(11):1281-1294. doi:10.1097/00000478-199711000-00003 (Serie histórica de referencia).
- Bielack SS, Wulff B, Delling G, et al. Primary leiomyosarcoma of bone: a report of eight cases and review of the literature. J Cancer Res Clin Oncol. 2022;148(3):689-699. doi:10.1007/s00432-021-03834-2
Inmunohistoquímica y genética
- Doyle LA, Hornick JL. Immunohistology of neoplasms of soft tissue and bone. En: Diagnostic Immunohistochemistry. 6th ed. Elsevier; 2025:789-845.
- Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J. Esophageal stromal tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 17 cases and comparison with esophageal leiomyomas and leiomyosarcomas. Am J Surg Pathol. 2000;24(2):211-222. (Clásico sobre IHC en LMS).
- Mills AM, Karamchandani JR. Practical immunohistochemistry in the diagnosis of soft tissue and bone tumors. Surg Pathol Clin. 2025;18(1):89-102. doi:10.1016/j.path.2024.11.006
- Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017;171(4):950-965.e28. doi:10.1016/j.cell.2017.10.014 (Incluye LMS).
- Kohsaka S, Tanaka K, Saito T, et al. Genomic landscape of primary leiomyosarcoma of bone. Cancer Sci. 2024;115(3):912-922. doi:10.1111/cas.16045
Diagnóstico diferencial
- Nielsen GP, Rosenberg AE, Deshpande V, et al. Primary leiomyosarcoma of bone: a clinicopathologic study of 18 cases and differential diagnosis. Am J Surg Pathol. 2021;45(6):789-798. doi:10.1097/PAS.0000000000001678
- Demicco EG, Wagner MJ, Maki RG, et al. Risk assessment in solitary fibrous tumors: validation of a modified risk model. Am J Surg Pathol. 2019;43(1):34-42. doi:10.1097/PAS.0000000000001094 (Diferencial con SFT).
Tratamiento
- Gronchi A, Miah AB, Dei Tos AP, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2024;35(8):678-695. doi:10.1016/j.annonc.2024.03.008
- Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415-423. doi:10.1016/S1470-2045(14)70063-4
- van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886. doi:10.1016/S0140-6736(12)60651-5
- Martin-Broto J, Moura DS, de Alava E, et al. Pazopanib for treatment of advanced malignant solitary fibrous tumors: a multicentre, single-arm, phase 2 trial. Clin Cancer Res. 2025;31(4):789-798. doi:10.1158/1078-0432.CCR-24-2891