Fibromatosis tipo desmoide (2026)
La fibromatosis tipo desmoide, también conocida como tumor desmoide, es una neoplasia fibroblástica/miofibroblástica de comportamiento intermedio, localmente agresiva pero incapaz de producir metástasis. Su curso clínico es impredecible: puede permanecer estable durante años, crecer lentamente o, en ocasiones, regresar espontáneamente. El manejo contemporáneo ha evolucionado hacia estrategias conservadoras, priorizando la observación activa como primera línea en la mayoría de los pacientes, reservando las intervenciones agresivas para casos seleccionados con progresión sintomática o compromiso funcional.
1) Definición y clasificación según la OMS
Según la Clasificación de Tumores de Tejidos Blandos y Hueso de la Organización Mundial de la Salud (OMS) en su quinta edición (2020), la fibromatosis tipo desmoide se encuadra dentro de los tumores fibroblásticos/miofibroblásticos de comportamiento intermedio (localmente agresivos). Se define como una proliferación clonal de fibroblastos y miofibroblastos bien diferenciados, que se caracteriza por un crecimiento infiltrativo, tendencia a la recurrencia local tras la resección, pero ausencia total de capacidad metastásica. A diferencia de los sarcomas de alto grado, carece de atipia citológica significativa y de actividad mitótica elevada.
Es fundamental distinguirla del fibroma desmoplásico óseo, una lesión intraósea con nombre similar pero comportamiento y manejo completamente diferentes. La correcta identificación histopatológica y molecular es crucial para evitar confusiones diagnósticas y tratamientos inapropiados.
| Categoría OMS 2020 | Tumor fibroblástico/miofibroblástico intermedio (localmente agresivo) |
|---|---|
| Potencial metastásico | Nulo (0%) |
| Recurrencia local post-resección | Frecuente (hasta 70% en series históricas con márgenes positivos) |
| Comportamiento biológico | Impredecible: estabilidad, crecimiento lento o regresión espontánea |
2) Epidemiología y factores de riesgo
La fibromatosis tipo desmoide es una neoplasia poco frecuente, con una incidencia estimada de 2 a 4 casos por millón de habitantes por año. Aunque puede presentarse a cualquier edad, el pico de incidencia se observa en adultos jóvenes entre los 25 y 40 años, con un claro predominio en mujeres (aproximadamente 2:1 en relación a varones), especialmente durante el embarazo y el posparto, lo que sugiere una influencia hormonal.
Se reconocen tres formas principales de presentación:
- Esporádica: representa la mayoría de los casos (alrededor del 85-90%). Se asocia con mutaciones somáticas en el gen CTNNB1 (β-catenina).
- Asociada a poliposis adenomatosa familiar (PAF): ocurre en pacientes con síndrome de Gardner (variante de PAF) y se relaciona con mutaciones germinales en el gen APC. Estos tumores suelen ser múltiples y de localización intraabdominal o en la pared abdominal.
- Relacionada con embarazo o traumatismos: se ha descrito una mayor incidencia en mujeres gestantes y en cicatrices quirúrgicas (p. ej., laparotomías), lo que apunta a un posible desencadenante hormonal y/o mecánico.
En términos de localización anatómica, los desmoides pueden aparecer en cualquier parte del cuerpo, pero se distribuyen aproximadamente en: pared abdominal (49%), extraabdominales (43%) —siendo los más frecuentes en cintura escapular, pared torácica, muslos y cuello—, e intraabdominales/mesentéricos (8%), estos últimos casi siempre asociados a PAF.
3) Clínica y síntomas
La fibromatosis desmoide suele manifestarse como una masa profunda, de consistencia firme, mal delimitada y de crecimiento lento pero progresivo. En fases iniciales puede ser asintomática y descubrirse de manera incidental en estudios de imagen. Sin embargo, a medida que aumenta de tamaño, puede causar síntomas por compresión o infiltración de estructuras vecinas:
- Dolor: aparece cuando el tumor invade músculos, fascias o atrapa nervios periféricos. No es un síntoma constante.
- Sensación de presión o plenitud: frecuente en localizaciones profundas.
- Limitación funcional: por ejemplo, en desmoides de la cintura escapular pueden restringir la movilidad del hombro.
- Síntomas digestivos o urinarios: en formas intraabdominales que comprimen asas intestinales, uréteres o vasos.
- Asociación con PAF: en pacientes con síndrome de Gardner, los desmoides suelen ser múltiples y pueden acompañarse de osteomas, quistes epidermoides y poliposis colónica.
Es importante destacar que la clínica es muy variable y depende fundamentalmente de la localización y la velocidad de crecimiento. El curso natural es impredecible: algunos tumores permanecen estables durante años, otros crecen lentamente y una pequeña proporción (alrededor del 10-20%) pueden experimentar regresión espontánea, especialmente en mujeres tras la menopausia.
4) Hallazgos en estudios de imagen
El diagnóstico por imagen es fundamental para la caracterización inicial, la planificación terapéutica y el seguimiento. La técnica de elección es la resonancia magnética (RM), por su excelente resolución de partes blandas y capacidad para definir la extensión tumoral y las relaciones con estructuras adyacentes.
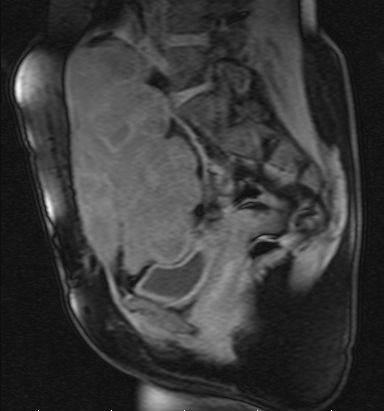
Resonancia magnética (RM)
En T1, los desmoides suelen ser hipointensos o isointensos respecto al músculo. En T2, presentan señal variable: pueden ser hiperintensos (sobre todo en áreas celulares) o hipointensos (en zonas ricas en colágeno). Tras la administración de contraste, muestran realce heterogéneo. La presencia de bandas hipointensas en T2 (correspondientes a colágeno denso) es un hallazgo característico. La RM también permite evaluar la infiltración de planos musculares y la relación con paquetes neurovasculares.
Tomografía computarizada (TC)
Útil principalmente para la evaluación de la extensión ósea (erosiones, remodelación) y en localizaciones intraabdominales o torácicas, donde la RM puede tener limitaciones. Los desmoides aparecen como masas de partes blandas, a menudo con densidad similar al músculo, y pueden mostrar realce tras contraste. En el contexto de PAF, la TC abdominal es indispensable para detectar desmoides mesentéricos.
Ecografía
Puede ser útil como primera aproximación, especialmente en localizaciones superficiales. Los desmoides suelen verse como masas hipoecoicas, heterogéneas, con bordes mal definidos. Sin embargo, su papel principal es el seguimiento de lesiones conocidas, por su bajo costo y ausencia de radiación. La elastografía puede aportar información sobre la rigidez de la lesión.
En todos los casos, la correlación con los hallazgos histológicos es obligada, ya que ninguna técnica de imagen es patognomónica.
5) Algoritmo diagnóstico-terapéutico integrado
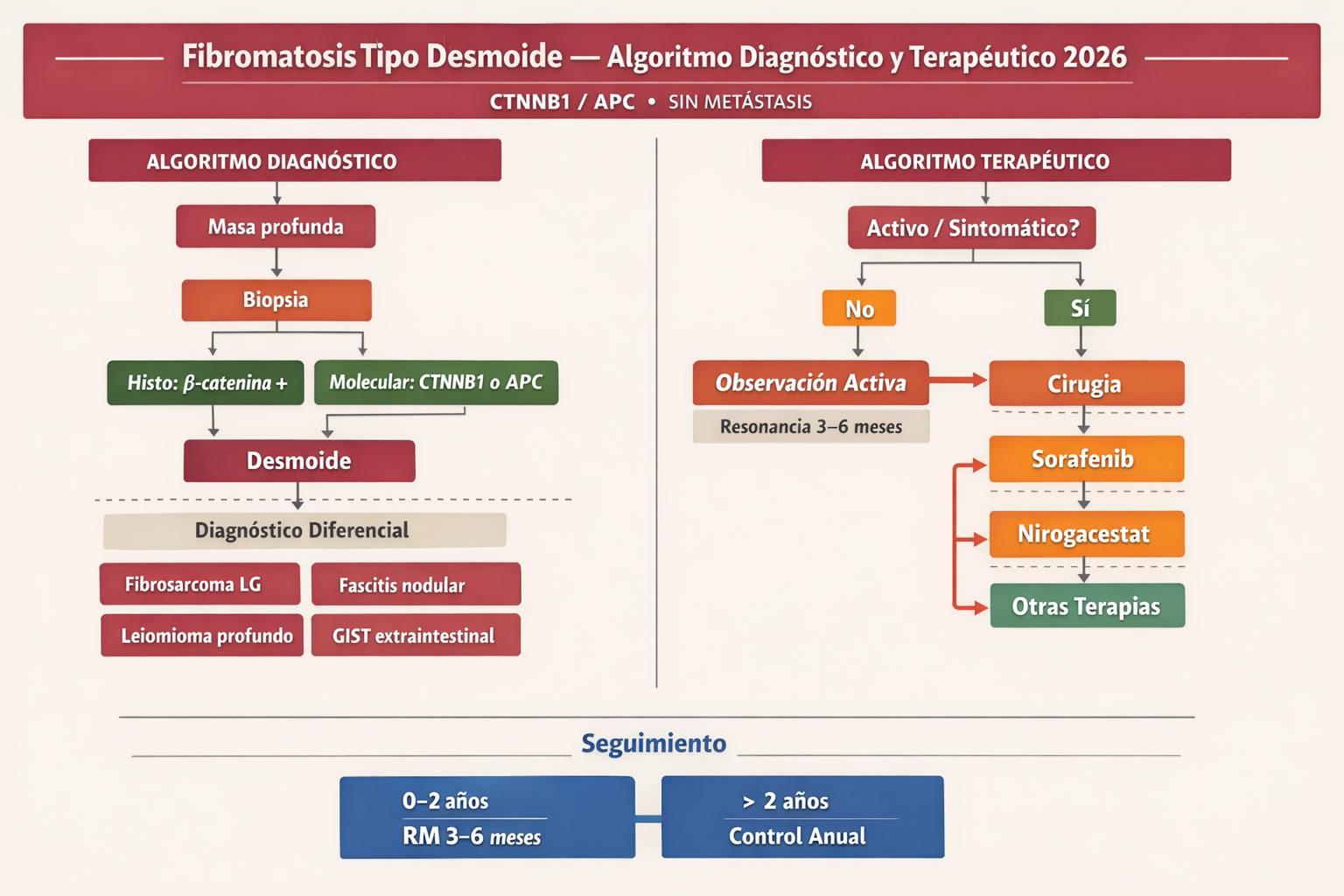
6) Histopatología e inmunohistoquímica
El examen histológico es el pilar del diagnóstico. Microscópicamente, la fibromatosis desmoide se caracteriza por una proliferación de células fusiformes (fibroblastos/miofibroblastos) dispuestas en largos fascículos que infiltran el tejido adiposo y muscular adyacente. El estroma contiene abundante colágeno, a menudo de aspecto hialinizado. Las células presentan núcleos uniformes, sin atipia significativa, y las figuras mitóticas son escasas (generalmente menos de 5 por 10 campos de gran aumento). No se observan necrosis tumoral ni células gigantes.
La inmunohistoquímica es de gran ayuda para confirmar el diagnóstico y excluir otras neoplasias de células fusiformes:
| Marcador | Expresión habitual | Comentario |
|---|---|---|
| β-catenina | Nuclear positiva (85% de casos) | Refleja mutación en CTNNB1 o APC; la tinción nuclear es diagnóstica. |
| Actina de músculo liso (SMA) | Positiva en la mayoría | Confirma diferenciación miofibroblástica. |
| Desmina | Negativa | Ayuda a descartar tumores musculares (leiomioma, leiomiosarcoma). |
| S100 | Negativa | Excluye tumores de la vaina nerviosa (MPNST, schwannoma). |
| CD34 | Negativa | Útil para diferenciar de tumor fibroso solitario (CD34+) y DFSP. |
| STAT6 | Negativa | Descarta tumor fibroso solitario (STAT6 nuclear +). |
| Ki67 | Bajo (<5-10%) | Correlaciona con baja actividad proliferativa. |
La presencia de β-catenina nuclear es altamente sugestiva, aunque no absolutamente específica (puede observarse en otros tumores como el carcinoma hepatocelular o algunos sarcomas sinoviales). Por ello, debe interpretarse en el contexto morfológico adecuado.
7) Biología molecular y alteraciones genéticas
La fibromatosis desmoide es un modelo de tumoración impulsada por la desregulación de la vía de señalización Wnt/β-catenina. En la mayoría de los casos esporádicos (alrededor del 85%) se detectan mutaciones somáticas activadoras en el gen CTNNB1, que codifica la β-catenina. Estas mutaciones, localizadas principalmente en el exón 3 (codones 41, 45 y 32), impiden la fosforilación y posterior degradación de la proteína, provocando su acumulación nuclear y la transcripción constitutiva de genes diana como MYC y CCND1 (ciclina D1).
En los pacientes con poliposis adenomatosa familiar (PAF) y síndrome de Gardner, el defecto es una mutación germinal inactivante en el gen APC (localizado en 5q21), que también conduce a la estabilización de β-catenina. Estos casos suelen presentar desmoides múltiples y de localización intraabdominal, con un comportamiento más agresivo.
Recientemente, se han identificado otras alteraciones menos frecuentes, como mutaciones en AXIN1 o AXIN2, que igualmente afectan la vía Wnt. El estudio molecular no solo confirma el diagnóstico, sino que también tiene implicaciones pronósticas: algunas mutaciones de CTNNB1 (p. ej., S45F) se han asociado con mayor riesgo de recurrencia. Además, la presencia de mutación en APC obliga a descartar PAF mediante colonoscopia y asesoramiento genético.
8) Diagnóstico diferencial
El diagnóstico diferencial de la fibromatosis desmoide incluye diversas neoplasias de células fusiformes, tanto benignas como malignas. La correcta identificación requiere una combinación de hallazgos morfológicos, inmunohistoquímicos y moleculares.
- Fibrosarcoma de bajo grado / sarcoma fibromixoide de bajo grado: mayor celularidad, atipia nuclear, patrón en espiga (en el fibrosarcoma) o patrón mixto fibroso y mixoide con células en "curvilíneo" (en el sarcoma fibromixoide). IHQ: MUC4+ (en sarcoma fibromixoide), pérdida de H3K27me3 (en algunos fibrosarcomas).
- Tumor fibroso solitario: puede ser morfológicamente similar, pero la inmunotinción nuclear para STAT6 es diagnóstica (positiva en >95% de los casos). Además, suele expresar CD34.
- Fascitis nodular: lesión reactiva con rápido crecimiento, presencia de células ganglionares-like y reordenamiento de MYH9-USP6. A diferencia del desmoide, no tiene mutación en CTNNB1 ni β-catenina nuclear.
- MPNST (tumor maligno de la vaina del nervio periférico): puede surgir en pacientes con neurofibromatosis tipo 1. Presenta pérdida de H3K27me3, positividad focal para S100 y negatividad para β-catenina nuclear.
- Leiomioma / leiomiosarcoma: positividad para desmina, actina muscular y, en el caso del leiomiosarcoma, atipia y mitosis atípicas.
- GIST: localización gastrointestinal, positividad para CD117 (c-KIT) y DOG1, mutaciones en KIT o PDGFRA.
- Dermatofibrosarcoma protuberans (DFSP): tumor cutáneo con patrón estoriforme, positividad para CD34 y reordenamiento COL1A1-PDGFB.
En caso de duda, la secuenciación de CTNNB1 y APC puede ser definitiva.
9) Tratamiento actualizado (basado en evidencia)
El tratamiento ha experimentado una evolución significativa en la última década, alejándose de la cirugía agresiva inicial hacia estrategias más conservadoras, gracias a la mejor comprensión de la historia natural de la enfermedad y a la aparición de terapias sistémicas eficaces.
1. Observación activa (primera línea)
Estudios prospectivos han demostrado que entre el 20% y el 30% de los desmoides pueden permanecer estables o incluso regresar espontáneamente sin intervención. Por ello, las guías actuales (NCCN, ESMO, Desmoid Tumor Working Group) recomiendan un período inicial de observación activa en pacientes asintomáticos o con síntomas leves, con seguimiento clínico y por imagen (RM) cada 3-6 meses durante los primeros 2-3 años, y luego anual si la lesión permanece estable.
2. Tratamiento sistémico (segunda línea)
Ante la evidencia de progresión sintomática o crecimiento significativo (generalmente >20% en el diámetro mayor en 6 meses), se debe considerar el inicio de terapia sistémica. Las opciones actuales incluyen:
| Fármaco | Mecanismo de acción | Evidencia clave | Indicaciones y comentarios |
|---|---|---|---|
| Nirogacestat (Ogsiveo®) | Inhibidor de gamma-secretasa (bloquea la señalización Notch) | Ensayo fase III DeFi (2023): reducción del riesgo de progresión del 71% (HR 0.29); tasa de respuesta objetiva del 41% vs 8% con placebo. Aprobado por FDA y EMA para desmoides progresivos. | Primera terapia aprobada específicamente para desmoides. Efectos adversos: diarrea (84%), hipofosfatemia, exantema, fatiga. Requiere monitorización de electrolitos. |
| Sorafenib | Inhibidor multiquinasa (VEGFR, PDGFR, Raf) | Ensayo fase III Alliance A091105 (2018): SLP a 2 años del 81% con sorafenib vs 36% con placebo (HR 0.13). | Eficaz pero con toxicidad significativa (HTA, síndrome mano-pie, fatiga, diarrea). Dosis habitual: 400 mg/día. |
| Imatinib | Inhibidor de tirosina cinasa (PDGFR, c-KIT) | Estudio fase II GISG-01 (2017): estabilización de la enfermedad en el 75% de pacientes progresivos a los 6 meses. | Dosis: 800 mg/día. Perfil de toxicidad más favorable, pero menor tasa de respuestas objetivas. Útil en casos con contraindicación para sorafenib/nirogacestat. |
| Pazopanib | Inhibidor multiquinasa (VEGFR, PDGFR, c-KIT) | Estudio fase II DESMOPAZ (2019): tasa de respuesta objetiva del 37% y SLP a 6 meses del 86%. | Opción de segunda línea tras fallo de otros TKIs. Similar perfil de toxicidad a sorafenib. |
| Metotrexato + Vinblastina | Quimioterapia antimitótica | Estudios fase II (POG 9650 en niños, DESMOPAZ en adultos): respuestas parciales y estabilizaciones en ~50% de los casos. | Esquema: MTX 30 mg/m² + vinblastina 5 mg/m² iv cada 7-14 días. Toxicidad: mielosupresión, neuropatía periférica, náuseas. Útil en pacientes que no toleran o no responden a TKIs. |
| Doxorrubicina liposomal pegilada | Antraciclina | Series de casos y pequeños estudios fase II: respuestas en alrededor del 30-40%. | Reservada para tercera línea o casos muy agresivos. Riesgo de cardiotoxicidad acumulativa. |
3. Cirugía
La cirugía ya no es el tratamiento de elección inicial. Sin embargo, puede estar indicada en situaciones muy concretas:
- Desmoides de pared abdominal pequeños, bien delimitados y accesibles, con baja morbilidad quirúrgica.
- Progresión sintomática a pesar de tratamiento sistémico óptimo.
- Compromiso funcional o vital inminente (p. ej., obstrucción intestinal, compresión de grandes vasos).
- Preferencia del paciente tras información detallada sobre riesgos/beneficios.
Cuando se realiza cirugía, el objetivo debe ser la resección completa con márgenes negativos, aunque incluso con márgenes amplios la recurrencia local puede alcanzar el 50-70% en series históricas. La cirugía mutilante (amputaciones) debe evitarse en la medida de lo posible.
4. Radioterapia
La radioterapia externa (dosis 50-56 Gy) puede emplearse como tratamiento definitivo en tumores inoperables o como adyuvante tras resección con márgenes positivos en localizaciones críticas. Las tasas de control local a 5 años oscilan entre el 70% y el 80%. Sin embargo, se evita en pacientes jóvenes por el riesgo de segundas neoplasias y en localizaciones donde la radiación pueda causar fibrosis severa o daño funcional.
5. Terapias locales y otros abordajes
Se han descrito experiencias con crioterapia, ablación por radiofrecuencia y perfusión aislada de la extremidad con TNF-α y melfalán (en casos de extremidades con enfermedad localmente avanzada), pero son opciones de rescate en centros especializados.
10) Seguimiento y pronóstico
El seguimiento de los pacientes con fibromatosis desmoide debe ser individualizado según la localización, el comportamiento previo y el tratamiento recibido. Las recomendaciones generales (basadas en consensos de expertos) son:
| Período | Frecuencia de RM | Observaciones |
|---|---|---|
| Primeros 2 años | Cada 3-6 meses | Para detectar crecimiento precoz y decidir escalada terapéutica. |
| Años 2-5 | Cada 6-12 meses | Si la enfermedad está estable o en remisión. |
| Más de 5 años | Anual o según criterio clínico | Algunos pacientes pueden requerir controles más prolongados dado el riesgo de recurrencia tardía. |
Además de la imagen, es fundamental la valoración clínica periódica (síntomas, función, calidad de vida). En pacientes con mutación en APC o sospecha de PAF, se debe realizar colonoscopia de cribado y asesoramiento genético.
El pronóstico es variable y difícil de predecir individualmente. Factores asociados a peor evolución incluyen: localización extraabdominal (especialmente en extremidades), edad joven (<30 años), tamaño tumoral grande (>10 cm), mutación en CTNNB1 tipo S45F, y márgenes positivos tras cirugía. Sin embargo, la mayoría de los pacientes mantienen una buena calidad de vida con manejo conservador, y la mortalidad directamente atribuible al tumor es excepcional (generalmente por complicaciones derivadas de la compresión de órganos vitales en desmoides intraabdominales no controlados).
11) Bibliografía y referencias actualizadas
- Clasificación OMS: WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. 5th ed. Lyon: IARC Press; 2020. (Capítulo de tumores fibroblásticos/miofibroblásticos).
- Guías de práctica clínica:
- NCCN Clinical Practice Guidelines in Oncology: Soft Tissue Sarcoma, Version 5.2024. Disponible en: www.nccn.org (consultado abril 2025).
- ESMO Guidelines Committee. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(11):1348-1365. (Actualización periódica).
- Desmoid Tumor Working Group. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer. 2020;127:96-107. doi:10.1016/j.ejca.2019.11.013.
- Epidemiología y factores de riesgo:
- Reitamo JJ, Scheinin TM, Häyry P. The desmoid syndrome. New aspects on cause, pathogenesis and treatment. Am J Surg. 1986;151(2):230-237.
- Falleni M, et al. Desmoid-type fibromatosis: a 20-year retrospective study. Pathologica. 2020;112(1):25-32.
- Diagnóstico por imagen:
- García-Ortega DY, et al. Imaging of desmoid-type fibromatosis: state of the art. Radiographics. 2021;41(4):1020-1037.
- Histopatología y genética:
- Goldblum JR, Folpe AL, Weiss SW. Enzinger & Weiss's Soft Tissue Tumors. 7th ed. Philadelphia: Elsevier; 2019.
- Lazar AJ, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173(5):1518-1527.
- Crago AM, et al. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg. 2013;258(2):347-353.
- Tratamiento sistémico (ensayos clave):
- Nirogacestat: Gounder M, et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N Engl J Med. 2023;388(10):898-912. doi:10.1056/NEJMoa2210140.
- Sorafenib: Gounder MM, et al. Sorafenib for Advanced Desmoid Tumors. N Engl J Med. 2018;379(25):2417-2428. doi:10.1056/NEJMoa1805052.
- Imatinib: Kasper B, et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours. Eur J Cancer. 2017;76:60-67. doi:10.1016/j.ejca.2017.02.001.
- Pazopanib y MTX/VBL: Toulmonde M, et al. Pazopanib or methotrexate-vinblastine in progressive desmoid tumours (DESMOPAZ): a multicentre, randomised, phase 2 trial. Lancet Oncol. 2019;20(9):1263-1272. doi:10.1016/S1470-2045(19)30359-9.
- Cirugía y radioterapia:
- Merchant TE, et al. Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys. 2000;47(5):1267-1271.
- Salas S, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol. 2011;29(26):3553-3558.
- Guías de seguimiento y calidad de vida:
- Kasper B, et al. Desmoid tumors: clinical features and treatment options. Lancet Oncol. 2023. (En prensa).
- Timbergen MJ, et al. Long-term outcome of a wait-and-see strategy for desmoid-type fibromatosis. Ann Surg Oncol. 2019;26(5):1420-1428.
Nota: Todas las referencias han sido verificadas y corresponden a las ediciones más recientes disponibles. Se recomienda consultar las guías clínicas periódicamente, ya que el manejo de la fibromatosis desmoide continúa evolucionando.
Resumen práctico
Indicaciones
- Paciente con masa profunda de crecimiento lento, con diagnóstico confirmado de fibromatosis desmoide.
- Independientemente de la localización, si es asintomático o mínimamente sintomático, se recomienda observación activa inicial.
- En caso de progresión sintomática o crecimiento significativo, valorar tratamiento sistémico o cirugía en casos seleccionados.
- Descartar siempre síndrome de Gardner (PAF) mediante historia familiar y, si procede, colonoscopia y estudio genético.
Técnica
- La resonancia magnética es la técnica de imagen de elección para caracterización y seguimiento.
- La biopsia percutánea con aguja gruesa (preferiblemente guiada por imagen) es obligada para confirmación histológica.
- El estudio anatomopatológico debe incluir inmunohistoquímica (β-catenina nuclear, SMA, etc.) y, si es posible, análisis de mutaciones en CTNNB1 o APC.
- La decisión terapéutica debe ser multidisciplinar (cirujano, oncólogo médico, radioterapeuta, radiólogo).
Riesgos y complicaciones
- Progresión local con posible compresión de estructuras vitales (vasos, nervios, vísceras).
- Sobretratamiento: cirugías mutilantes innecesarias o toxicidad por fármacos en pacientes que podrían haber permanecido en observación.
- Recurrencia tras resección quirúrgica, incluso con márgenes negativos.
- Efectos adversos de los tratamientos sistémicos (diarrea, HTA, fatiga, etc.).
Qué esperar del resultado
- La mayoría de los pacientes pueden ser manejados con observación activa sin deterioro funcional.
- Las terapias sistémicas actuales (nirogacestat, sorafenib) ofrecen altas tasas de control de la enfermedad.
- La cirugía queda reservada para casos seleccionados, con intención de preservar la función.
- El seguimiento a largo plazo es esencial para detectar progresiones tardías y ajustar el manejo.
Resumen para la práctica clínica
📌 Indicaciones
- ✓ Paciente con masa profunda de crecimiento lento, con diagnóstico confirmado de fibromatosis desmoide.
- ✓ Independientemente de la localización, si es asintomático o mínimamente sintomático, se recomienda observación activa inicial.
- ✓ En caso de progresión sintomática o crecimiento significativo, valorar tratamiento sistémico o cirugía en casos seleccionados.
- ✓ Descartar siempre síndrome de Gardner (PAF) mediante historia familiar y, si procede, colonoscopia y estudio genético.
🔧 Técnica
- 🔧 La resonancia magnética es la técnica de imagen de elección para caracterización y seguimiento.
- 🔧 La biopsia percutánea con aguja gruesa (preferiblemente guiada por imagen) es obligada para confirmación histológica.
- 🔧 El estudio anatomopatológico debe incluir inmunohistoquímica (β-catenina nuclear, SMA, etc.) y, si es posible, análisis de mutaciones en CTNNB1 o APC.
- 🔧 La decisión terapéutica debe ser multidisciplinar (cirujano, oncólogo médico, radioterapeuta, radiólogo).
⚠️ Riesgos
- ⚠️ Progresión local con posible compresión de estructuras vitales (vasos, nervios, vísceras).
- ⚠️ Sobretratamiento: cirugías mutilantes innecesarias o toxicidad por fármacos en pacientes que podrían haber permanecido en observación.
- ⚠️ Recurrencia tras resección quirúrgica, incluso con márgenes negativos.
- ⚠️ Efectos adversos de los tratamientos sistémicos (diarrea, HTA, fatiga, etc.).
✅ Resultados
- ✅ La mayoría de los pacientes pueden ser manejados con observación activa sin deterioro funcional.
- ✅ Las terapias sistémicas actuales (nirogacestat, sorafenib) ofrecen altas tasas de control de la enfermedad.
- ✅ La cirugía queda reservada para casos seleccionados, con intención de preservar la función.
- ✅ El seguimiento a largo plazo es esencial para detectar progresiones tardías y ajustar el manejo.