Última revisión:
Cordoma
El cordoma es un tumor maligno poco frecuente, de crecimiento lento, que se origina en restos de la notocorda embrionaria y se localiza casi exclusivamente en la línea media del esqueleto axial. Históricamente considerado de bajo grado, hoy sabemos que su comportamiento es localmente agresivo, con alta tasa de recidiva local y potencial metastásico. El reto principal es: (1) su diagnóstico, a menudo tardío por localización profunda; (2) lograr una resección quirúrgica completa (difícil por su relación con estructuras vitales); y (3) el manejo de recidivas y enfermedad avanzada.
1) Definición y conceptos clave
Según la Clasificación de Tumores de la OMS (5ª edición, 2020), el cordoma es una neoplasia maligna que muestra diferenciación notocordal. Surge de vestigios de la notocorda (estructura embrionaria precursora del núcleo pulposo) que no involucionaron completamente. No se origina en el núcleo pulposo del disco intervertebral adulto; este es un concepto erróneo ya superado.
Lo que importa en la práctica clínica actual:
- • Localización axial estricta: Sacro (50-60%), base del cráneo/clivus (25-35%) y vértebras móviles (15%).
- • Marcador clave: Expresión nuclear del factor de transcripción brachyury, esencial en el diagnóstico diferencial.
- • Comportamiento agresivo: Aunque crece lentamente, es infiltrativo, recidivante y puede metastatizar (principalmente a pulmón, hueso, hígado y piel).
2) Epidemiología y genética
Datos epidemiológicos actuales
El cordoma es un tumor muy raro. Representa aproximadamente el 1-4% de todos los tumores óseos primarios y alrededor del 20% de los tumores primarios de la columna vertebral. Afecta principalmente a adultos, con un pico entre la 5ª y 7ª décadas, aunque puede aparecer a cualquier edad, incluidos niños y adolescentes (cordoma pediátrico, a menudo de clivus, con comportamiento potencialmente más agresivo). Existe un predominio masculino con una relación de aproximadamente 2:1.
En cuanto a la localización, el sacro y coccíx concentran el 50-60% de los casos, la base del cráneo y clivus el 25-35%, y las vértebras móviles el 15% restante (siendo más frecuente en la columna cervical, especialmente C2, y lumbar).
Genética y factores de riesgo
La gran mayoría de los cordomas son esporádicos. Existe una forma familiar ligada a la duplicación del gen brachyury (gen T) en el cromosoma 6q27, que confiere un riesgo aumentado. Mutaciones en genes como LYST y PIK3CA también se han descrito. No hay evidencia de vínculo con traumatismos previos.
3) Presentación clínica
Los síntomas son insidiosos, inespecíficos y dependen críticamente de la localización, lo que explica el retraso diagnóstico frecuente (a menudo de más de un año). La siguiente tabla resume las presentaciones clínicas según la localización:
| Localización | Síntomas más frecuentes | Complicaciones / Compresión |
|---|---|---|
| Sacro/Coccíx | Dolor lumbar/sacro/glúteo crónico (90%), peor en sedestación. Estreñimiento, tenesmo rectal. | Masa presacra palpable (tacto rectal). Compresión de raíces sacras (ciatalgia, déficits sensitivo-motores, síndrome de cauda equina). Incontinencia urinaria/fecal. |
| Base del Cráneo (Clivus) | Cefalea suboccipital/retroorbitaria. Diplopía (visión doble) por afectación del VI par (nervio abducens). | Parálisis de pares craneales (VI, III, IV, V, IX, X). Compresión del tronco encefálico. Obstrucción del flujo de LCR (hidrocefalia). |
| Vértebras Móviles | Dolor axial localizado. Rigidez. Tortícolis (en cervicales). | Mielopatía por compresión medular (debilidad, alteración de marcha). Radiculopatía. Fractura patológica. Inestabilidad vertebral. |
4) Señales de alarma y derivación
Indicaciones ABSOLUTAS para derivación urgente a una Unidad de Sarcomas/Tumores de Columna:
- Dolor axial (lumbar/sacro/cervical) persistente (>4-6 semanas), progresivo, no mecánico (no mejora con reposo) o nocturno.
- Masa palpable presacra o en región glútea detectada en exploración física o tacto rectal.
- Síntomas neurológicos progresivos de cualquier tipo: radiculopatía, paresia, alteraciones sensitivas, disfunción esfinteriana (vesical/anal).
- Hallazgo radiológico de una lesión lítica, expansiva, de la línea media en sacro, clivus o vértebra, especialmente con masa de partes blandas asociada.
- Ante la sospecha, la resonancia magnética (RM) es la prueba de imagen de primera línea.
Regla de oro: En el contexto de dolor axial persistente con hallazgos radiológicos atípicos, siempre descartar patología tumoral. La derivación precoz es crucial para el pronóstico.
5) Diagnóstico por imagen (RX, TC, RM)
Radiografía (RX) simple
Tiene un papel limitado hoy en día, pero a menudo es la primera prueba. Puede mostrar una lesión lítica, expansiva y destructiva en la línea media, con borramiento de planos grasos y, en ocasiones, calcificaciones intratumorales. En el sacro, se puede observar destrucción de los agujeros sacros anteriores.
Tomografía computarizada (TC)
Es excelente para valorar la arquitectura ósea: destrucción lítica, expansión, esclerosis reactiva y, sobre todo, calcificaciones intralesionales (60-70% de los casos). Es fundamental para la planificación quirúrgica y la biopsia guiada. La TC de tórax/abdomen forma parte del estadiaje para detectar metástasis.
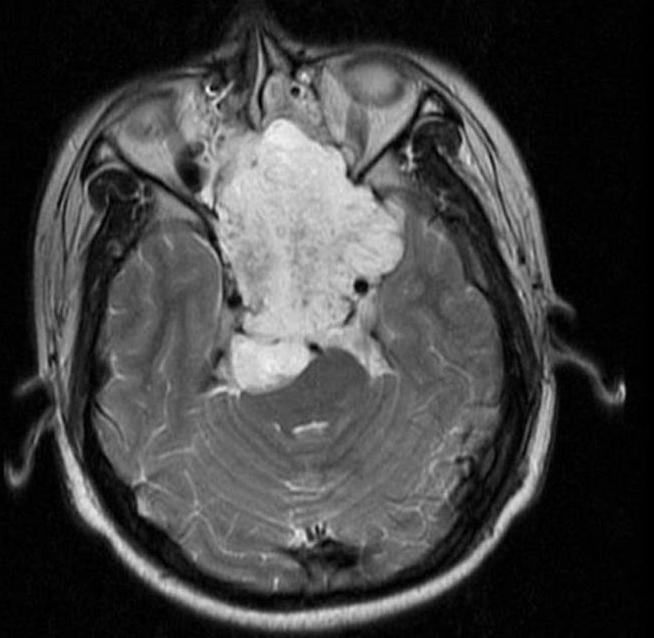
Resonancia magnética (RM)
Es la técnica de imagen más importante y definitiva. La señal es hipointensa en T1 (a menos que haya hemorragia) e hiperintensa en T2 por su alto contenido acuoso/mixoide. Se realza de forma heterogénea e intensa con contraste (gadolinio). La RM define con precisión la extensión intramedular, el componente de partes blandas, la relación con estructuras neurovasculares críticas (médula, raíces, vasos, vísceras) y la respuesta al tratamiento.
Pruebas obsoletas que ya NO se usan: La mielografía y la gammagrafía ósea (que puede ser negativa, "fría", o mostrar captación periférica) han sido reemplazadas por la RM y la TC.
6) Biopsia y diagnóstico histológico
PRINCIPIO FUNDAMENTAL: La biopsia de una lesión sospechosa de cordoma (o cualquier tumor de columna/pelvis) DEBE ser planificada y realizada por o en estrecha coordinación con el equipo quirúrgico que llevará a cabo la resección definitiva. Una biopsia mal planteada puede contaminar planos anatómicos y convertir una resección potencialmente curativa en una imposible.
La biopsia percutánea con aguja gruesa (tru-cut) guiada por TC es el estándar. El estudio histológico requiere:
- H&E: Arquitectura lobulada. Células epitelioides en cordones o nidos dentro de una abundante matriz mixoide. Presencia de células fisalíferas (células grandes con vacuolas citoplasmáticas que desplazan el núcleo, "en sello de anillo").
- Inmunohistoquímica (IHQ) DIAGNÓSTICA:
- Brachyury (nuclear): POSITIVO. Marcador altamente específico y de gran sensibilidad. Clave para diferenciarlo de metástasis de adenocarcinoma o condrosarcoma.
- Citoqueratinas (CK AE1/AE3, CAM5.2, CK19): POSITIVO. Confirma diferenciación epitelial.
- Proteína S-100: POSITIVO (en ≈80% de los casos).
- EMA: Variablemente positivo.
7) Patología y subtipos (OMS 2020)
La 5ª edición de la OMS clasifica los cordomas en tres subtipos principales, con implicaciones pronósticas y terapéuticas:
| Subtipo | Frecuencia | Características clave | Pronóstico / Comportamiento |
|---|---|---|---|
| 1. Convencional (Clásico) | ≈70-80% | Arquitectura lobulada, células fisalíferas, matriz mixoide. Brachyury (+). | Crecimiento lento pero localmente agresivo. Potencial metastásico (≈30-40% a largo plazo). |
| 2. Desdiferenciado | <5% | Zonas de cordoma convencional adyacentes a un sarcoma de alto grado de estirpe no condroide. Brachyury se pierde en el componente sarcomatoso. | MUY AGRESIVO. Mayor tasa de metástasis y peor supervivencia. Requiere quimioterapia sistémica. |
| 3. Poorly Differentiated (PDC) (NUEVO en OMS 2020) |
≈10% (más en niños) | Células pequeñas, redondas, azules, con pérdida de la arquitectura lobular. Positiva para brachyury y pérdida de SMARCB1 (INI1). Asociado a alteraciones en SMARCB1. | Comportamiento agresivo. Similar al desdiferenciado. Alta tasa de metástasis. Más común en base de cráneo pediátrica. |
Nota sobre el "subtipo condroide": Este término, usado en clasificaciones antiguas, ha sido abandonado por la OMS 2020. Se consideraba una variante de crecimiento lento que simulaba cartílago, pero el marcador brachyury permite su correcta clasificación como cordoma convencional.
8) Marcadores moleculares y genética
El descubrimiento de la expresión de brachyury ha revolucionado el diagnóstico. Es un factor de transcripción clave en el desarrollo de la notocorda. Su expresión nuclear detectada por IHC es altamente específica para cordoma, diferenciándolo de sus miméticos (carcinoma metastásico, condrosarcoma mixoide, paracordoma).
Aplicaciones prácticas de la genética
- Diagnóstico: Brachyury es la prueba de confirmación.
- Dianas terapéuticas: Se investigan fármacos dirigidos contra vías relacionadas con brachyury.
- Pronóstico: La pérdida de SMARCB1 (INI1) define el subtipo Poorly Differentiated y se asocia a peor pronóstico.
- Consejo genético: En casos raros familiares, se puede estudiar la duplicación del gen brachyury.
9) Tratamiento multidisciplinar
El manejo DEBE realizarse en centros de referencia con equipos multidisciplinares (cirujanos de columna/neurocirujanos/cirujanos pélvicos, oncólogos radioterápicos, oncólogos médicos, radiólogos, patólogos).
1. Cirugía
Objetivo: Resección en bloque (en bloc) con márgenes quirúrgicos amplios (R0). Es el factor pronóstico más importante para la supervivencia libre de enfermedad.
- Sacro: Sacrectomía parcial o total. La preservación de las raíces S2-S3 de un lado puede mantener la continencia.
- Base del cráneo: Accesos endoscópicos endonasales extendidos (EEA) o craneotomías. La resección completa es muy difícil.
- Vértebras: Espondilectomía total en bloque (si es posible).
2. Radioterapia
Papel crucial por la frecuente imposibilidad de cirugía R0. Se usa como adyuvante postquirúrgica (si márgenes son R1/R2) o primaria en tumores irresecables.
- Estándar de oro: Radioterapia de intensidad modulada con partículas pesadas (protones o iones carbono). Permite administrar dosis altas (70-74 Gy) con un gradiente de caída muy abrupto, preservando tejidos sanos críticos (médula, tronco encefálico).
- Fotones: IMRT/VMAT de alta precisión es una alternativa si no hay acceso a partículas.
- Radiocirugía (SRS): Para recidivas pequeñas o metástasis.
3. Terapia Sistémica (para enfermedad metastásica/irresecable)
Históricamente quimiorresistente. Nuevas opciones han cambiado el panorama:
- Inhibidores de la tirosina quinasa: Imatinib (diana: PDGFR-β) y Sorafenib han mostrado actividad y estabilización de la enfermedad en un porcentaje significativo de pacientes. A menudo se usan de por vida como terapia crónica.
- Inmunoterapia: En estudio, con resultados variables.
- Quimioterapia clásica: De elección únicamente para el subtipo desdiferenciado, que se trata como un sarcoma de alto grado.
10) Pronóstico y seguimiento
Pronóstico
La supervivencia global a 5 años ronda el 70-80%, pero a 10 años cae al 40-50% debido a las recidivas tardías. La supervivencia libre de progresión es más baja. Factores de mal pronóstico: cirugía no R0, tamaño tumoral grande (>8-10 cm), subtipo desdiferenciado o Poorly Differentiated, localización en base del cráneo y edad avanzada.
Esquema de seguimiento recomendado
- Años 1-3: RM local y TC tórax/abdomen cada 6 meses.
- Años 4-10: RM local y TC tórax/abdomen anualmente.
- >10 años: RM local cada 1-2 años (riesgo de recidiva muy tardía).
El seguimiento debe ser de por vida debido al riesgo de recurrencia décadas después.
11) Algoritmo de manejo práctico

Resumen para la práctica clínica
Indicaciones
- ✓ Dolor axial (sacro/lumbar/cervical) persistente (>4-6 semanas) y no mecánico.
- ✓ Masa palpable presacra o en la base del cráneo.
- ✓ Hallazgo en imagen (RM/TC) de lesión lítica, expansiva, en la línea media del esqueleto axial.
- ✓ Síntomas neurológicos progresivos (radiculopatía, pares craneales, incontinencia).
Técnica
- 🔧 RM de columna completa/región afectada con contraste (técnica de elección). TC para valoración ósea y planificación.
- 🔧 Biopsia percutánea guiada por TC, PLANIFICADA por el equipo quirúrgico definitivo.
- 🔧 Estudio histológico con IHC obligatoria: Brachyury (nuclear), Citoqueratinas, S-100.
- 🔧 Tratamiento en centro de referencia: Cirugía en bloque (R0) + RT con partículas (protones) si es posible.
Riesgos
- ⚠️ Recidiva local (alta probabilidad si cirugía no R0). Puede ser tardía (>10 años).
- ⚠️ Metástasis a distancia (pulmón, hueso, hígado) en aproximadamente un tercio de los casos.
- ⚠️ Secuelas neurológicas y funcionales importantes de la cirugía radical (déficits, incontinencia).
- ⚠️ Progresión de enfermedad localmente avanzada o metastásica.
Resultados
- ✅ La resección quirúrgica completa (R0) es el principal factor pronóstico favorable.
- ✅ La radioterapia con partículas (protones/carbono) mejora significativamente el control local.
- ✅ Los TKIs (imatinib/sorafenib) pueden estabilizar la enfermedad avanzada durante años.
- ✅ Seguimiento de por vida por riesgo de recidiva muy tardía.
12) Bibliografía actualizada
Soft Tissue and Bone Tumours. 5th ed. IARC; 2020.
Best practices for the management of local-regional recurrent chordoma. Ann Oncol. 2017;28(6):1230-1242.
Chordoma: the entity. Biochim Biophys Acta. 2014;1846(2):655-669.
Bone Cancer (versión vigente).
The role of brachyury in chordoma pathogenesis and targeted therapy. Cancer Lett. 2023;558:216-224.
Cordoma: guía actualizada
Diagnóstico · Subtipos · Tratamiento multidisciplinar · Seguimiento