... Condromas ...
-
Osteocondromas
-
- Condromatosis múltiple (encondromatosis, enfermedad de Ollier) y síndrome de Maffucci
-
- Condroma de tejidos blandos
Condromas
- Condroma solitario encondroma)
- Condroma perióstico
Diagnóstico diferencial de los condromas
Condromas
El condroma es una lesión del cartílago hialino maduro que puede ser uno de dos tipos:
(1) encondroma que se localiza centralmente dentro de la cavidad medular del hueso
(2) condroma perióstico, yuxtacortical o cortical que se levantan en o bajo el periostio.
La lesión se caracteriza por tejido cartilaginoso maduro, benigno.
La transformación maligna ocurre raramente.
El condroma se ha informado que se da en un 25 % (616 de 2421) de tumores benignos del hueso y 12 % (616 de 5274) de tumores del hueso primarios demostrado con biopsia. El condroma se descubre a lo largo de la vida. Los pacientes con lesiones múltiples normalmente están entre diez y treinta años, con un predominio máximo que ocurre durante la tercera década de vida. No hay ninguna predilección del sexo.
El encondroma es bastante común en los huesos pequeños de las manos y pies, con 58% (ochenta y nueve de 153) de las lesiones que ocurre en estas áreas. De hecho, el encondroma es el tumor primario más común encontrado en los huesos de la mano. El encondroma asintomático normalmente se encuentra, incidentalmente, en las radiografías. El encondroma atípico se caracteriza así debido a su asociación con una historia de dolor. A veces se ve una fractura en el área del encondroma, quizás debido a debilitad previa del hueso o simplemente como un hallazgo incidental.
Ocasionalmente, un paciente tiene encondromatosis múltiple, o enfermedad de Ollier. Un paciente que tiene encondromas múltiples y angiomas de los tejidos blandos tiene el síndrome de Maffucci. La transformación sarcomatosa de los encondromas puede ocurrir en asociación con estos dos síndromes.
El condroma periosteal es un tumor cartilaginoso raro, benigno que se levanta de los tejidos periósticos. Es el equivalente perióstico del encondroma medular. Los nombres adicionales para esta lesión son condroma parosteal y condroma yuxtacortical. Los signos y síntomas del condroma perióstico son similares a los del encondroma solitario. El dolor es el síntoma más común, y la lesión normalmente se encuentra incidentalmente. Raramente, si alguna vez, se convierte a una lesión maligna.
Condroma Solitario (encondroma)
El
encondroma
es
una
lesión
del
cartílago
hialino
maduro,
benigna
y
asintomática,
es
el
tumor
cartilaginoso
de
hueso
que
se
da,
más
a
menudo,
en
adolescentes
o
adultos
jóvenes;
Es una lesión del cartílago intramedular localizada en el centro de la metáfisis.
El
tumor
intramedular
se
desarrolla
en
la
metáfisis
adyacente
y
puede,
Es el resultado del fracaso de osificación endocondral normal debajo del platillo de crecimiento y representa una displasia del platillo de crecimiento central.
Si
el
proceso
displásico
ocurre
en
el
platillo
de
crecimiento
lateral,
el
tumor
resultante
se
llama
osteocondroma.
La proliferación displásica cartilaginosa bajo el pericondrio produce el condroma periosteal.
Localización:
Normalmente
afecta
a
huesos
tubulares
pequeños
de
manos
o
pies
(40-65%).
Es
el
tumor
primario
más
común
encontrado
en
las
manos.
El
encondroma,
normalmente
es
asintomático,
y
es
un
hallazgo
incidental
radiográfico.
El
encondroma
atípico
se
llama
así
por
presentar
una
historia
de
dolor. Es
una
causa
frecuente
de
fractura
patológica.
Lo
más
frecuentemente
es
que
involucre
la
falange
proximal
seguida
por
la
falange
medial,
y
los
metacarpianos. También
puede
afectar
húmero
proximal
y
la
diáfisis
femoral.
Otros sitios que puede afectar son los huesos
largos, como fémur, húmero, peroné y costillas.
Al contrario que el condrosarcoma, el encondroma de los huesos planos (iliaco, escápula y esternón) es relativamente raro, así como el localizado en la columna, especialmente en los cuerpos vertebrales.
Riesgo
de
transformación
maligna:
El
encondroma
normalmente
se
pone
latente
en
la
madurez,
y
<
2%
de
encondromas
asintomáticos
solitarios
se
transforman
en
condrosarcoma.
Los
encondromas
de
huesos
largos
tienen
el
riesgo
más
alto
de
transformación
maligna.
En la encondromatosis, el riesgo de transformación maligna es aproximadamente 10-25%.
En la enfermedad de Maffucci (encondromas múltiple y hemangiomas) el riesgo está cercano 100%.
Los hallazgos radiográficos de transformación maligna incluyen la actividad biológica aumentada, como evidencia del aumento del festoneado endosteal, la fractura patológica, o el aumento de tamaño.
El rastreo óseo del área puede o no indicar aumento de actividad y no es una manera fiable de evaluar la transformación maligna.
La RM es ocasionalmente útil para evaluar el componente no mineralizado de la lesión y su aparente agresividad.
Si los estudios de imagen y la situación clínica indican potencial para la transformación maligna, es obligatoria la biopsia.
El
encondroma
no
causa síntomas
a
menos
que
haya
fractura
patológica.
Aunque pueden aparecer a cualquier edad, al mayoría se presenta entre la segunda y cuarta década.
Histología:
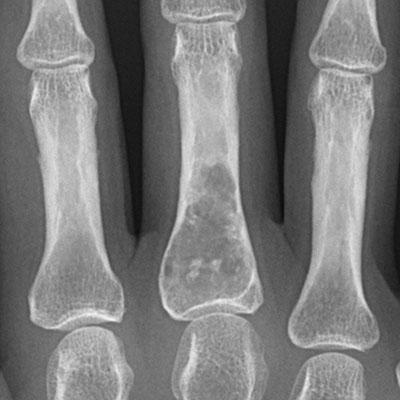
El encondroma es una lesión larga, oval que se localiza centralmente en la porción tubular del hueso. Normalmente tiene groseramente y típicamente una apariencia lobulada y bien delimitada (los lóbulos son de tamaños diferentes y separados por septos pequeños de tejido fibroso). La calcificación de la lesión puede o no estar presente. La expansión de la corteza circundante es rara a menos que la lesión ocurre en un hueso pequeño, como los huesos de las manos o pies, o en el peroné.
Los encondromas están compuestos de células cartilaginosas benignas. Los signos de posible transformación maligna, como el aumento de la celularidad y las células binucleadas ocasionales, son compatibles con un diagnóstico benigno, sobre todo en las manos y pies, pero la diferenciación entre el encondroma y los condrosarcomas grado-1 pueden ser histológicamente bastante difíciles. La citología que ofrece el encondroma y el condrosarcoma grado-1 a menudo se imbrican. El tamaño de los núcleos, el número de células por unidad de área, el número de condrocitos binucleados, y el número de mitosis, son características importantes que deben evaluarse. La distinción entre el encondroma y el condrosarcoma de bajo grado no se hace ,estrictamente, en base al criterio histológico; el criterio radiográfico también es útil. Debe haber una comunicación íntima entre el cirujano, el patólogo, y el radiólogo para formular el último diagnóstico.
Lesión
radiolucente
central
bien
definida,
mínima
condensación
de
margen
óseo.
Durante
la
fase
activa
en
el
adolescente,
la
lesión
puede
crecer
lentamente.
En
los
niños,
la
cortical
está
normalmente
delgada
y
la
lesión
es
radiolucente.
Posteriormente
aparecen
las
calcificaciones
intralesionales.
Estas
calcificaciones,
generalmente
tiene
forma
de
puntos,
anillos,
y
arcos
La cortical está adelgazada de modo fusiforme y tiene unos márgenes internos festoneados, que refleja el patrón de crecimiento lobular del cartílago.
La frecuente apariencia nubosa da una pista de la naturaleza condroide de la lesión.
No hay ninguna reacción del periostio.
En la fase latente, el tejido cartilaginoso puede calcificar con el modelo punteado difuso. Cuando la lesión madura, evoluciona a un margen reactivo.
La RNM y la TAC pueden ayudar a delimitar el tumor y la localización más exacta en el hueso. Como en la mayoría de las lesiones cartilaginosas y fibrosas la RNM muestra el encondroma con baja o intermedia señal en T! y alta en T2. Las calcificaciones se ven vacías o como focos de baja señal. Con gadolinio la imagen se realza y ayuda a diferenciar la de una lesión no cartilaginosa.
Rastreo
óseo:
Demuestra la captación del radioisótopo en el margen, relacionado con la actividad de lesión.
Mientras
hay
captación
moderada
en
la
fase
activa,
habrá
también
alguna
actividad
en
la
fase
latente.
Tratamiento:
Los encondromas solitarios asintomáticos pueden seguirse no operativamente con series radiográficas.
El
pronóstico
para
el
encondroma
benigno
es
excelente.
Si
los
encondromas
solitarios
o
múltiples
se
vuelven
sintomáticos
o
empieza
a
crecer,
pueden
requerir
la
biopsia
para
descartar
malignidad.
Atención
a
la
tríada
terrible:
dolor,
aumento de
la
captación
de
radioisótopo
en
el
scan
óseo,
y
los
cambios
destructivos
en
la
radiografía.
-
Fractura
patológica:
En la mayoría de los casos la fractura consolida con tratamiento cerrado.
Después de la curación de la fractura, se requiere curetaje e injerto óseo, ya que la resolución espontánea del encondroma después de la curación de la fractura no es probable que ocurra.
El
riesgo
bajo
de
repetición
de
encondroma
y
el
condrosarcoma
de
bajo
grado
en
las
extremidades:
80
pacientes
siguieron
durante
2-25
años.
[HC]
el
[Bauer]
[et]
al.
[Acta]
[Orthop].
[Scand].
[Vol]
66.
el
[p283-288].
Condroma Perióstico
Discusión:
El
condroma perióstico o yuxtacortical es un tumor
cartilaginoso
que
aparece
en
la
superficie
cortical
profundo
al
periostio
produciendo
una masa
cartilaginosa
de
base
ancha
que
puede
extenderse
en
los
tejidos
blandos.
A menudo se desarrolla después de la adolescencia (en contraste con el osteocondroma).
Persiste como una masa de cartílago maduro con calcificación u osificación (en contraste con el condrosarcoma).
El
tumor
no
infiltra
el
tejido
blando
adyacente
pero
puede
aumentar
de
tamaño.
Dos tercios de estas lesiones aparecen en la metáfisis, y el otro tercio en la diáfisis.
Más
del
50%
de
estos
tumores
se
encuentra
en
la
corteza
lateral
de
húmero
proximal
justo
proximalmente
la
inserción
del
músculo
deltoides;
Otras lesiones se dispersan uniformemente por fuera los huesos largos.
Manifestaciones
clínicas:
Los
pacientes
pueden
quejarse
de
dolor
en
el
sitio
del
tumor.
A
menudo
puede
palparse
una
masa
dura
unida
al
hueso.
Radiografías:
Consiste
en
una
pequeña
masa
superficial
(<3
centímetro)
que
aparece
como
una
radiolucencia
ovalada
o
un
defecto
oblongo
en
la
periferia
de
la
corteza
subyacente
La lesión se refuerza por un anillo, distinguible de reacción cortical.
El
tumor
tiene
pequeñas
o
ninguna
calcificación. (en
contraste
con
el
condrosarcoma).
Ocasionalmente
hay
calcificaciones
intralesionales
y
la
reacción
del
periostio
es
mínima.
- diagnóstico diferencial radiográfico:
-
el
osteocondroma
en
los
pacientes
más
jóvenes.
-
el
condrosarcoma
yuxtacortical.
-
el
sarcoma
parosteal
y
periosteal.
-
TAC:
- La TAC se usa para demostrar la magnitud de la implicación cortical e identificar la densidad del cartílago.
Histología:
La
visión
con
baja
intensidad
muestra
masas
hialinas
lobuladas
bien
circunscritas.
La
lesión
normalmente
es
hipocelular
y
bien
circunscrita.
Se
pueden
ver
áreas
aisladas
de
celularidad
aumentada.
La lesión está compuesta de cartílago benigno pero parece más activa que un encondroma.
Esta
lesión
puede
confundirse
con
el
condrosarcoma.
Tratamiento:
La mayoría de estas lesiones se presentan en fase 2 y requieren excisión marginal en bloque o la excisión amplia, que incluye la cortical subyacente, para prevenir la recurrencia, es el tratamiento de elección.
El riesgo de repetición después de la excisión marginal en bloque es <10%, y la excisión más agresiva, la quimioterapia adyuvante o radioterapia no están indicadas.
Condromatosis Múltiple.
(Encondromatosis)
Las encondromatosis
son un grupo de enfermedades
caracterizadas
por
múltiples
encondromas,
que
generalmente
implican
la
metáfisis
y
la
diáfisis de huesos en crecimiento, principalmente de las
extremidades inferiores, no obstante también se han reportado en
sitios menos habituales como son: pelvis, costillas, base del cráneo,
septum nasal, senos
paranasales y tráquea.
Cualquier parte del esqueleto puede ser afectada..
Dichas lesiones son generalmente benignas, sin embargo su principal complicación es la transformación maligna en condrosarcomas.
La enfermedad de Ollier y el síndrome de Maffucci son las
encondromatosis mejor descritas y estudiadas en la literatura, sin
embargo no son las únicas, existe una gran variedad de estos
síndromes, entre los que también destaca la exostosis múltiple
familiar.
Enfermedad de Ollier
Fue descrita en el año 1900 por el cirujano francés Leopold
Ollier, como una enfermedad no hereditaria, de etiología desconocida,
sin predominio de género, que se presenta entre el segundo y décimo
año de vida. Por estos motivos,
muchos
autores
la
consideran
más
como
una
displasia
ósea
que
como
una
neoplasia. Se caracteriza por la
presencia unilateral de discondroplasia y múltiples encondromas,
localizados en los huesos largos de las extremidades y asociados a
anormalidades esqueléticas, secundarias al acortamiento de las
extremidades, escoliosis, fracturas en terreno patológico y
seudoartrosis. otros sitios menos frecuentemente afectados son el ileo
seguidos
de
falanges,
metacarpianos
y
metatarsianos.
En
muchos
menos
casos
la
lesión
afecta
a
huesos
faciales,
cráneo,
columna
carpo
y
tarso.
Síndrome de Maffucci
El síndrome de Maffucci es un trastorno congénito no hereditario, caracterizado por encondromatosis y angiomatosis de los tejidos blandos: viscerales o mucocutaneos. La hemangiomatosis puede situarse en la piel y en el tejido subcutáneo. Los hemangiomas son generalmente cavernosos y pueden ser unilaterales o bilaterales, localizados o extensos. Las lesiones esqueléticas en el síndrome de Maffucci tienen la misma distribución que en la enfermedad de Ollier, especialmente en los huesos de las manos y los pies.
Su nombre se debe al patólogo italiano Angelo Maffucci, quien lo describió en el año 1881. En este síndrome los encondromas son más frecuentes en los huesos cortos de las manos y pies y son diagnosticados durante la edad preescolar. El diagnóstico diferencial de estas dos entidades es difícil, debido a sus similitudes, por lo que se ha propuesto que son diferentes grados de expresión de una misma patología. El origen de estas enfermedades aún es desconocido, pero se cree que se debe a una displasia mesodérmica congénita o a una anomalía cromosómica aún no identificada.
Los signos y síntomas del síndrome de Maffucci pueden ser detectables al nacer, aunque generalmente no se hacen evidentes hasta alrededor de los 5 años de edad. Los encondromas se desarrollan cerca de los extremos de los huesos, cerca del cartílago de crecimiento y con frecuencia dejan de formarse después de que los individuos afectados dejen de crecer. (en la edad adulta temprana). Como resultado de las deformidades óseas asociadas con el síndrome de Maffucci , las personas con este trastorno generalmente tienen estatura baja y músculos subdesarrollados.
Exostosis múltiple
Una tercera entidad es la exostosis múltiple, la cual, a diferencia
de los síndromes anteriores, tiene un patrón de herencia ya conocido:
autosómico dominante. Se han identificado dos genes supresores
tumorales, involucrados en esta enfermedad: EXT1 y EXT2, localizados
en los cromosomas 8q24 y 11p11-p12, respectivamente. Actualmente se
sugiere la existencia de un tercer gen (EXT3) en el cromosoma 19p,
sin embargo aún no ha sido identificado.Su cuadro clínico se
caracteriza por el desarrollo de dos o más encondromas en las
epífisis de los huesos largos, principalmente en la articulación
femorotibial. Se presenta durante la primera década de la vida, con
una edad media de diagnóstico a los 3 años de edad. A diferencia de
otras encondromatosis, una vez que se cierran los cartílagos de
crecimiento, se detiene la formación de nuevos encondromas. Las
malformaciones musculoesqueléticas más frecuentes son: el
acortamiento del cúbito con arqueamiento del radio (39-60%),
crecimiento asimétrico de las extremidades (10-50%), baja estatura
(37-44%), angulación en varo o valgo de la rodilla (8-33%) y
deformidad de tobillo (2-54%).
Presentación
clínica
Las
manifestaciones
clínicas
son
abultamientos
nudosos
de
los
dedos
o
la
discrepancia
longitud
de
los
miembros.
Los
niños
afectos empiezan
a
claudicar
al
segundo
año
de
vida
así
como
a
presentar
una
discrepancia
en
longitud
(los
huesos
afectados
son
más
cortos
por
la
afectación
de
la
placa
de
crecimiento).
El
dolor
es
raro
y
cuando
se
presenta
generalmente
es
debido
a
una
fractura
patológica.
La
enfermedad
de
Ollier
a
menudo
se
detiene
en
la
pubertad
pero
puede
ocasionalmente,
continuar
progresando.
La
complicación
más
frecuente
de
la
enfermedad
de
Ollier y el síndroome de Maffucci
es
la
transformación
maligna
de
uno
o
más
encondromas
a
condrosarcomas. Schakovich
cree
que un
30%
de
pacientes
con
enfermedad
de
Ollier y el síndroome de Maffucci
evolucionan
a
condrosarcoma,
pero
Jaffe
y
Mirra
creen
que
esta
proporción
es
del
50%.
No
obstante,
como
los
pacientes
con
encondromatosis
pueden
desarrollar
centenares
de
encondromas,
el
riesgo
estadístico
de
que
una
lesión
evolucione
a
condrosarcoma
es
el
mismo
que
es
el
de
un
encondroma
solitario.
Diagnóstico
El diagnóstico de las encondromatosis se realiza bajo la sospecha
clínica y se apoya en estudios de imagen. No existen estudios
específicos para su diagnóstico. Los pacientes parecen normales al
nacimiento, sin embargo, durante su infancia presentan deformidades
esqueléticas secundarias al desarrollo de los encondromas o la
presencia de fracturas patológicas. Las personas con estas
enfermedades, por lo general tienen una vida normal, y la
inteligencia no se ve afectada. El grado de su deterioro físico
depende de sus deformidades esqueléticas individuales, pero en la
mayoría de los casos no tienen limitaciones importantes en sus
actividades. Los hallazgos clínicos más comunes son: acortamiento de
las extremidades, lesiones tumorales y fracturas de las áreas
afectadas, la enfermedad puede progresar hasta la discapacidad del
paciente o autolimitarse al final de la adolescencia. Los datos
sugestivos de malignidad de un encondroma son dolor y crecimiento
progresivo.
Estudios
de
imagen
La radiografía simple es el estudio inicial en la evaluación de los encondromas que generalmente se encuentran localizados en la diáfisis de los huesos largos y se observan como lesiones ovaladas, lobuladas y bien delimitadas.
Las radiografías de la en condromatosis de las manos y los pies son características. Se ven masas radiolucentes de cartílago con focos de calcificación, parecidos a los de los encondromas solitarios, que deforman severamente los huesos. Los encondromas en esta localización pueden ser intracorticales y periosteales. Algunas veces sobresalen de la diáfisis de los huesos cortos o largos, y asemejan a los osteocondromas. Pero estas proporciones nunca poseen una cubierta cartilaginosa de un tallo óseo. En los huesos largos se ven bandas radiolucentes que se extienden desde el platillo decrecimiento hasta la diáfisis. La coalescencia de encondromas metafisarios, a menudo provoca un agrandamiento asimétrico de los huesos largos.
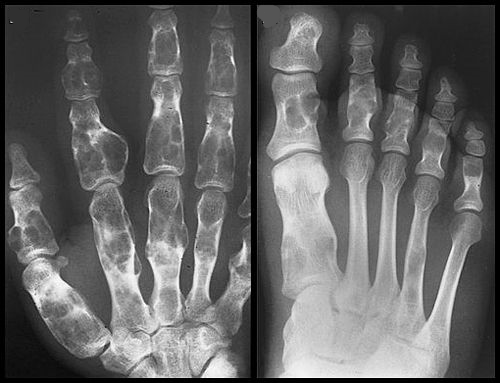
En el síndrome de Maffucci radiográficamente se observan múltiples flebolitos calcificados junto con las típicas alteraciones de la encondromatosis.
La TAC permite valorar la formación de matriz ósea y los órganos intraabdominales, mientras que la RMN valora la extensión a tejidos blandos, permitiendo la identificación de hemangiomas y tumores en el SNC. Una vez identificado un encondroma con sospecha de malignidad, lo recomendado es tomar una biopsia trucut guiada por imagen.
Histología
Las
lesiones
de
la
encondromatosis
son
indistinguibles
del
encondroma
solitario.
Desde el punto de vista histopatológico, el diagnóstico diferencial
de los encondromas y los condrosarcomas de bajo grado es difícil, ya
que ambas entidades presentan hipercelularidad y atipia leve, por lo
que el diagnóstico diferencial se apoya en la agresividad del cuadro
clínico y en los estudios de imagen.
Tratamiento
Los encondromas tienen un riesgo mayor de malignidad en comparación
a cuando se presentan de
forma aislada y requieren tratamiento sólo en el caso de
sintomatología o sospecha de malignidad, en cuyo
caso el tratamiento de elección es la resección quirúrgica de la
lesión.
Diagnóstico diferencial de los Condromas
Radiológico
El
principal
diagnóstico
diferencial
del
encondroma
solitario,
sobre
todo
cuando
se
localiza
en
un
hueso
largo
y
muestra
una
importante
calcificación
es
con
el infarto
óseo
medular.
Las
diferencias
se
basan
en
los
márgenes
lobulados
del
encondroma,
en
las
calcificaciones
punteadas
sin
matriz,
y
la
ausencia
de
esclerosis
periférica
fibro-ósea
que
se
observa
en
el
infarto.
Los
depósitos
de
mineral
también
tienen
tendencia
a
mostrar
una
distribución
más
central,
y
las
calcificaciones
a
menudo
tienen
una
marcada
apariencia
punteada.
La
distribución
del
infarto
óseo
es
generalmente
el
fémur
proximal
y
distal
y
en
la
tibia
proximal,
esta
distribución
no
es
frecuente
en
el
encondroma.
El
problema
más
importante
es
diferenciar
un
encondroma
grande
de
un condrosarcoma
de
bajo
grado.
El
condrosarcoma
de
bajo
grado
en
estadio
precoz
se
localiza
en
la
cortical.
El
tamaño
de
la
lesión
también
ayuda
en
el
diagnóstico.
Las
lesiones
mayores
de
4
cm
son
sugestivas
de
ser
malignas.
En
las
lesiones
más
avanzadas
la
destrucción
de
la
cortical
y
la
masa
de
tejidos
blandos
sugieren
malignidad.
También
es
importante
distinguir
la transformación
sarcomatosa en
pacientes
con
encondromas
múltiples
y
en
la
enfermedad
de
Ollier.
Radiográficamente
se
demuestra
radiolucencia
intralesional
y
destrucción
cortical
asociada
con
una
masa
de
tejidos
blandos.
Si el encondroma, particularmente en huesos tubulares cortos, se extiende a la superficie articular de hueso, puede entrar en el diagnóstico diferencial con el tumor de células gigantes. Este no tiene calcificaciones, aunque en algunos encondromas estas calcificaciones pueden no ser visibles. Además el tumor de células gigantes carece de borde esclerótico. En los huesos tubulares largos el encondroma raramente se extiende en la superficie articular del hueso.
El encondroma puede recordar un quiste óseo solitario, aunque este último raramente ocurre en los huesos tubulares cortos.
En
los
encondromas
de
las
falanges
distales
de
las
manos
que
no
contenga
calcificación
puede
confundirse
con
un quiste
epidermoide.
Una
encondroma
intracortical
puede
confundirse
con
un osteocondroma,
no
obstante
la
distribución
de
la
matriz
mineral
es
diferente
en
ambas
entidades.
El
encondroma
protuberante
que
es
una
encondroma
exofítico
de
un
hueso
largo
o
de
las
costillas,
nace
de
la
cavidad
medular
pero
penetre
en
la
cortical,
formando
una
prominencia
exofítica
en
la
superficie
del
hueso
y
puede
asemejarse
a
una lesión
superficial (osteoma,
osteocondroma,
condroma
periostal,
miositis
osificante).
El
condroma
periostal
deberá
ser
diferenciado
de
las lesiones
subperiósticas (ganglión
periostal,
sarcoma
de
Ewing
periosteal),
así
como
de
la miositis
osificante y
la calcinosis
tumoral.
El
condroma
periostal
grande
puede
confundirse
con
un osteocondroma
sesil.
La
lesión
de
condroma
periosteal
está
separada
de
la
porción
medular
del
hueso
por
una
cortical
invertida.
La
cortical
del
osteocondroma
se
fusiona
con
la
cortical
del
hueso
huésped,
y
hay
continuidad
entre
la
médula
del
hueso
huésped
y
la
exostosis.
La
TAC
la
ayuda
al
distinguir
a
estas
dos
enfermedades.
Patológico
El mayor problema es distinguir entre encondroma y condrosarcoma de bajo grado. En la mayoría de los casos no hay problemas de distinción pero si la lesión no es celular, y particularmente si afecta a los huesos largos, el patólogo debe tener cuidado de no confundir un encondroma de un condrosarcoma de bajo grado.